
X Chimie PC 2011
| Thème de l'épreuve | Le dioxyde de titane et la photocatalyse. Utilisation de l'acide de Mosher en synthèse organique. |
| Principaux outils utilisés | mélanges binaires, thermochimie, orbitales moléculaires, cinétique, chimie organique |
| Mots clefs | acide de Mosher, dioxyde de titane, photocatalyse, acide bleu 80, AB80, amphidinolide T1 |
Corrigé
:👈 gratuite pour tous les corrigés si tu crées un compte
👈 l'accès aux indications de tous les corrigés ne coûte que 1 € ⬅ clique ici
👈 gratuite pour tous les corrigés si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
👈 gratuite pour ce corrigé si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Énoncé complet
(télécharger le PDF)












Rapport du jury
(télécharger le PDF)








Énoncé obtenu par reconnaissance optique des caractères
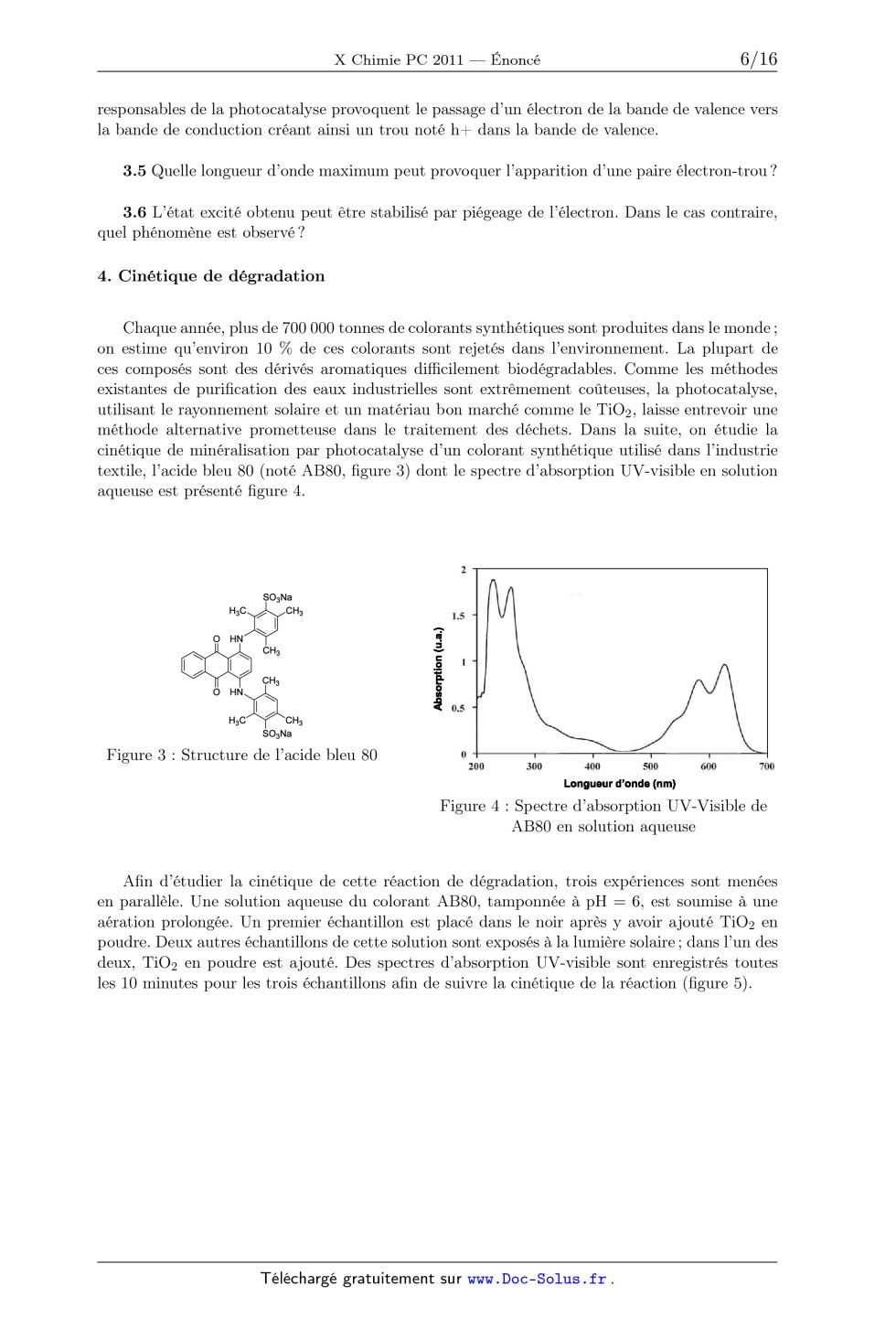
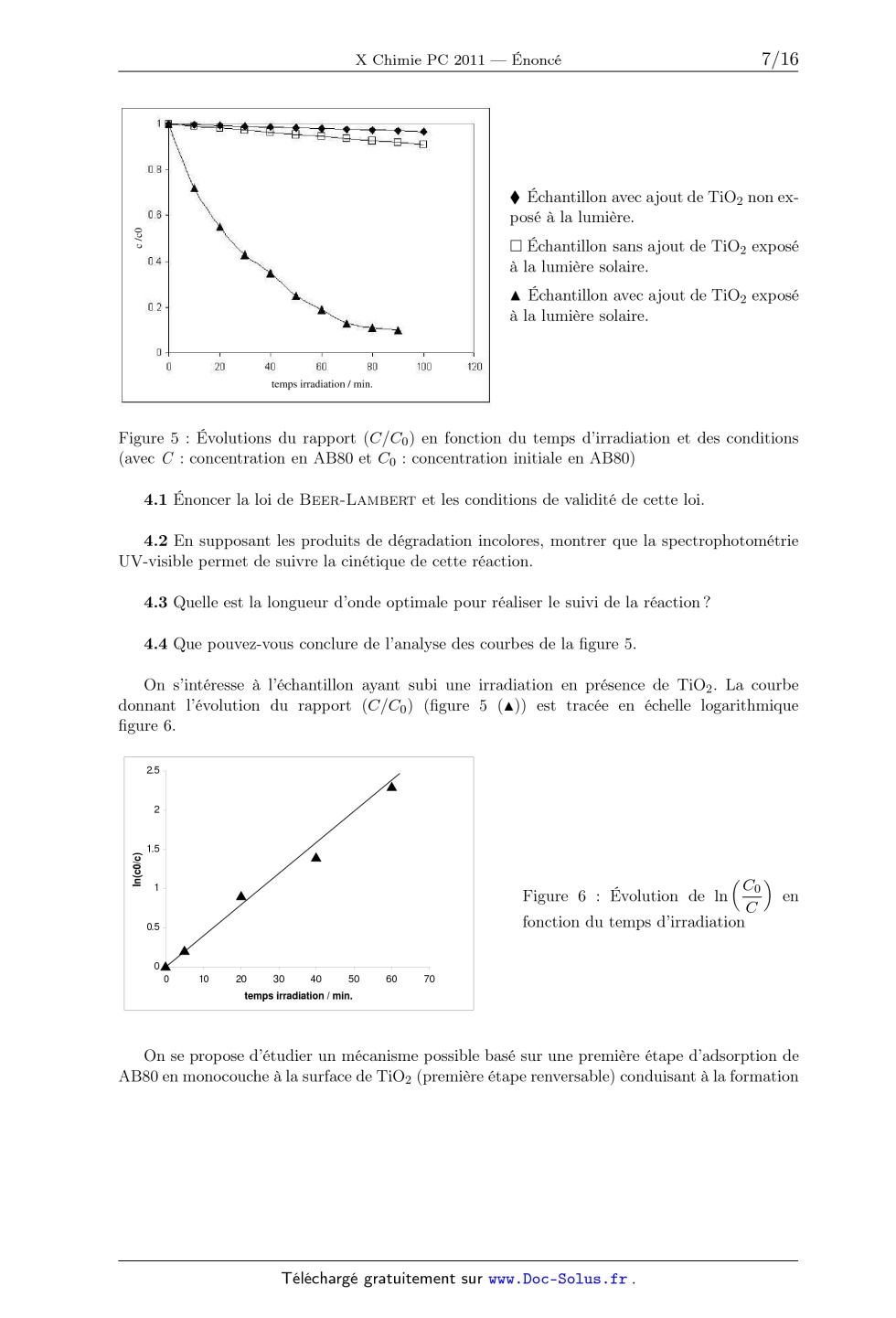
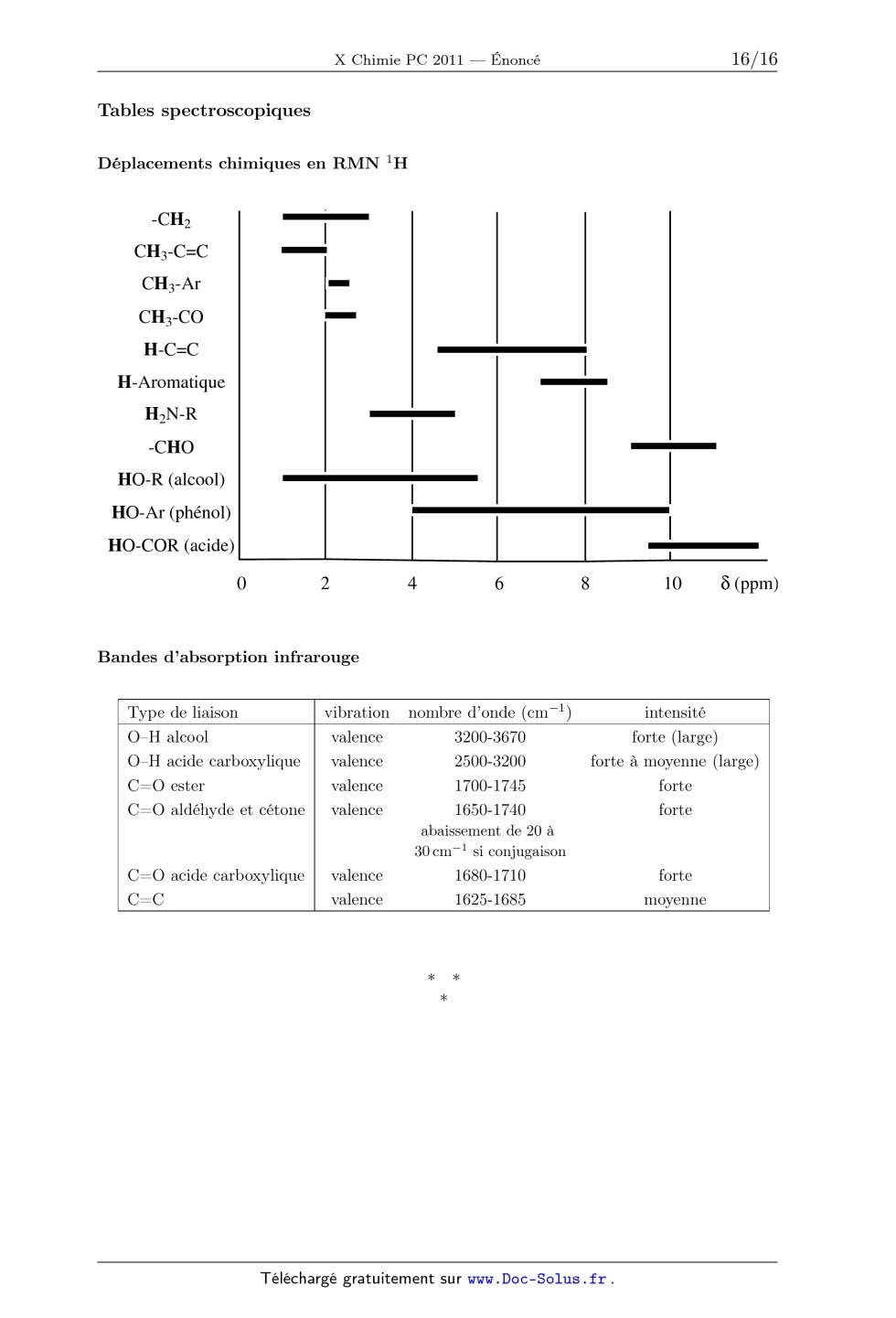
ÉCOLE POLYTECHNIQUE ÉCOLES NORMALES SUPÉRIEURES ÉCOLE SUPÉRIEURE DE PHYSIQUE ET DE CHIMIE INDUSTRIELLES CONCOURS D'ADMISSION 2011 FILIÈRE PC COMPOSITION DE CHIMIE (XEULC) (Durée : 4 heures) L'utilisation des calculatrices n'est pas autorisée pour cette épreuve. Premier problème Le dioxyde de titane et la photocatalyse La photocatalyse permet de réaliser la décomposition de polluants organiques par oxydation de la matière sous l'action du rayonnement électromagnétique (principalement UV) en utilisant un matériau semi-conducteur. Ainsi, le dioxyde de titane TiO2 est utilisé pour favoriser la minéralisation des molécules organiques (transformation de ces composés en CO2 , H2 O, et - éventuellement SO2- 4 et NO3 . . . ). Cette technique trouve de nombreuses applications dans la vie quotidienne comme, par exemple, les vitres autonettoyantes, la purification de l'eau ou de l'air . . . La photocatalyse est principalement une catalyse hétérogène, il est donc essentiel de maîtriser la pureté et de connaître l'état de surface du matériau utilisé. Données : · · · · · 1 eV = 1,6×10-19 J. Constante de Planck : h = 6,62×10-34 J · s. Constante d'Avogadro : NA = 6,02×1023 mol-1 . Constante des gaz parfaits : R = 8, 31 J · K-1 · mol-1 Vitesse de la lumière dans le vide : c = 3 · 108 m · s-1 . 1. Obtention de TiO2 pur Le TiO2 pur est obtenu par oxydation de TiCl4 selon la réaction suivante : TiCl4 (g) + O2 (g) = TiO2 (s) + 2 Cl2 (g) TiCl4 est lui-même préparé à partir de TiO2 , extrait du milieu naturel, dont la principale impureté est la silice (SiO2 ), laquelle peut nuire aux propriétés photocatalytiques du matériau. Une fois les oxydes transformés en dérivés chlorés (TiCl4 et SiCl4 ), il est possible d'obtenir TiCl4 pur par distillation fractionnée, étudiée ici en supposant que le mélange TiCl4 /SiCl4 se comporte comme un mélange idéal aussi bien en phase liquide qu'en phase vapeur (Les gaz seront assimilés à des gaz parfaits). Les fractions molaires respectives des composés TiCl4 et SiCl4 en phase liquide 1 seront notées xTi et xSi et les fractions molaires en phase gazeuse yTi et ySi . Les enthalpies standard de vaporisation de TiCl4 et SiCl4 seront notées vap HoTiCl4 et vap HoSiCl4 . Elles seront supposées indépendantes de la température. Le tableau 1 présente les coordonnées de différents points du diagramme d'équilibre liquidevapeur pour le mélange binaire TiCl4 /SiCl4 sous une pression P pression standard, P = 1 bar. / C xSi ySi 57 1 1 65 0,76 0,98 75 0,54 0,94 89 0,33 0,86 100 0,22 0,76 108 0,15 0,65 115 0,10 0,54 125 0,05 0,33 132 0,02 0,15 136,5 0 0 Tableau 1 : Coordonnées de différents points du diagramme d'équilibre liquide-vapeur pour le mélange binaire TiCl4 /SiCl4 sous la pression P o . 1.1 Donner l'allure du diagramme d'équilibre liquide-vapeur de ce mélange binaire idéal. 1.2 Quel est le composé le plus volatil ? 1.3 Pour les deux composés du mélange sous une pression totale P , exprimer les constantes des équilibres physiques en fonction de l'activité des constituants présents. 1.4 Exprimer les lois de variation de ces deux constantes en fonction de la température, des enthalpies standard de vaporisation et des températures d'ébullition des corps purs. 1.5 En déduire les expressions de ln Ä yTi ä xTi et ln Ä ySi ä xSi . 1.6 Donner alors les expressions de xTi et yTi en fonction de la température, des enthalpies standard de vaporisation, des températures d'ébullition des corps purs et des pressions P et P . Soit un mélange constitué initialement de 15 mol de SiCl4 et 85 mol de TiCl4 à = 20 C dans une enceinte fermée maintenue à la pression P . 1.7 Déterminer sans calcul : 1.7.1 l'état physique initial du mélange ; 1.7.2 la température de début d'ébullition du mélange, et la composition de la première bulle de vapeur ; 1.7.3 la composition et les quantités de matière de chacune des deux phases si le mélange est porté à 115 C ; 1.7.4 la température à laquelle s'achève la vaporisation. Une cascade de distillations élémentaires appliquée au mélange SiCl4 /TiCl4 permet d'obtenir en tête de colonne une phase enrichie en l'un des deux constituants, mais le rendement est très faible. Pour l'améliorer, il est nécessaire de recycler une partie du liquide obtenu en tête de 2 colonne, ce que nous examinons maintenant. On notera A le composé le plus volatil et B le composé le moins volatil. Sur la colonne présentant deux plateaux (voir Figure 1), on définit : . an : la quantité de matière recueillie après condensation en tête de colonne (distillat) pendant un temps dt, . a:AD : la fraction molaire du composé A dans la phase liquide du distillat, . dn; : la quantité de matière de liquide refluant et arrivant sur le plateau z' pendant un temps dt, . dnv : la quantité de matière de vapeur issue du plateau (75 -- 1) pendant un temps dt, . a:Ai : la fraction molaire du composé A dans la phase liquide sur le plateau z' , . yAi : la fraction molaire du composé A dans la phase vapeur sur le plateau Z. condenseur nD, XD Plateau 2 n| Plateau 1 V mélange à distiller bou...eur Figure 1 : Principe de fonctionnement de la colonne a distiller On étudie le fonctionnement de la colonne en supposant que sur chaque plateau, l'équilibre thermodynamique est réalisé. Au condenseur, la phase vapeur est totalement condensée, une partie est récupérée et l'autre partie est renvoyée dans la colonne. On suppose que la pression est uniforme et constante de 1 bar a l'intérieur de la colonne et que la quantité de matière de chaque constituant présente sur chaque plateau est constante. On souhaite obtenir :cAD = 0,98, cette valeur est applicable pour les questions 1.8 a 1.15. 1.8 Indiquer la valeur de yA2 et en déduire a:A2 ainsi que la température T2 du plateau 2. 1.9 En écrivant la conservation de la quantité totale de matière, donner une relation entre dn... dnl et an. 1.10 En écrivant la conservation de la quantité de matière en constituant A pour le plateau 2, donner une relation entre yA1, :cA2, a:AD, dnl et an. 1.11 Exprimer alors yA1 en fonction de a:A2, 56 AD et dn;/an. 1.12 Que devient cette expression pour an/an infiniment grand (soit a reflux total)? 1.13 Calculer yA1 pour dn;/an : 1. En déduire a:A1. 1.14 Calculer alors la fraction molaire du composé A dans la vapeur présente dans le bouilleur. En déduire la fraction molaire du composé A dans le liquide présent dans le bouilleur. 1.15 Ce dispositif a-t-il permis de purifier TiCl4 ? 2. Détermination de la surface spécifique du matériau La surface spécifique d'un matériau est définie comme la surface de sites actifs par gramme de matériau utilisé, exprimée en m2 · g-1 . Afin de la déterminer, une masse m = 4,0 g de poudre de TiO2 est introduite dans une enceinte sous pression de diazote N2 . Une fois l'équilibre d'adsorption de N2 sur TiO2 atteint, la pression de diazote régnant dans l'enceinte est notée P. L'étude de cet équilibre est réalisée en tenant compte des hypothèses suivantes : une seule molécule peut être adsorbée par site d'adsorption ; l'adsorption conduit à la formation d'une monocouche ; les molécules adsorbées n'interagissent pas entre elles. Les échanges gaz-surface sont schématisés de la façon suivante : N2 + K S S N2 Figure 2 : Échanges gaz-surface où S- représente un site libre, S-N2 une molécule N2 adsorbée à la surface. La fraction des sites occupés est notée . On définit Ntot comme la quantité de matière de sites d'adsorption de TiO2 . 2.1 En considérant que l'activité des sites libres ou occupés est assimilable à leur fraction, exprimer en fonction de K, P et P . Une première série d'expériences réalisées pour différentes valeurs P de pression en diazote permet de déterminer la surface spécifique . On relève ainsi pour chaque P , la quantité de matière de diazote adsorbée, notée Nads , et on trace la droite 1/Nads en fonction de 1/P . 2.2 Préciser l'expression de l'ordonnée à l'origine et de la pente de cette droite. 2.3 Sachant que l'ordonnée à l'origine de la droite vaut 2000 mol -1 et qu'une molécule de diazote a un encombrement de = 1, 6 × 10-19 m2 , évaluer la surface spécifique de la poudre de TiO2 considérée. 2.4 De la même manière, on obtient une surface spécifique de = 560 m2 · g-1 pour un gel de silice sous pression de diazote. Commenter. Afin de connaître les grandeurs de réaction du phénomène d'adsorption, on réalise une seconde expérience en faisant varier la température de l'enceinte. En relevant la pression nécessaire à une adsorption identique à ces différentes températures, on détermine l'enthalpie isostérique d'adsorption, notée ads H . ads H correspond à l'enthalpie standard d'adsorption pour un recouvrement de la surface fixé, c'est-à-dire à fixée. 4 2.5 Montrer que le tracé de la droite ln P en fonction de 1/T permet d'accéder à ads H . 2.6 Pour = 0,8, la pente de cette droite vaut -1, 7 × 103 K. Calculer la valeur de ads H correspondante. Commenter. 2.7 Expliquer comment déterminer l'entropie isostérique d'adsorption. En réalité, les échanges gaz-surface schématisés précédemment sont en compétition avec un processus dissociatif de la molécule de diazote au contact de la surface. 2.8 Dans le cas limite d'une dissociation complète du diazote, illustrer par un schéma analogue à la Figure 2, les échanges gaz-surface se produisant au moment de l'adsorption, en faisant figurer la constante K' (par analogie avec K). 2.9 Exprimer alors , en fonction de K , P et P . 2.10 Commenter ce résultat en comparant à la loi obtenue en 2.1. 3. Principe simplifié de la photocatalyse Pour étudier l'effet d'un rayonnement sur un matériau, il est nécessaire de connaître la distribution énergétique des électrons dans les orbitales moléculaires de ce matériau. 3.1 Représenter le diagramme d'énergie des orbitales moléculaires de la molécule de dihydrogène H2 ainsi que la répartition des électrons dans ces orbitales dans l'état fondamental de cette molécule. Considérons un enchaînement linéaire d'un grand nombre n d'atomes d'hydrogène régulièrement espacés. Il est possible de déterminer les orbitales moléculaires de cet enchaînement par la méthode de Hückel simple. Les énergies Ep associées à ces orbitales moléculaires (1 6 p 6 n) sont de la forme suivante : Ep = + 2 cos Ä p ä n+1 dans laquelle représente l'énergie d'un électron occupant une orbitale 1s d'un atome d'hydrogène et l'intégrale de résonance provenant de l'interaction entre les orbitales atomiques appartenant à deux atomes voisins. 3.2 Montrer que les énergies sont comprises entre deux valeurs que l'on précisera. 3.3 Montrer alors que lorsque n tend vers l'infini, les niveaux Ep forment un quasi-continuum d'énergie, appelée bande d'énergie. 3.4 Décrire la répartition des électrons dans cette bande à 0 K. On supposera que le modèle des bandes est applicable au dioxyde de titane. On établit que, dans ce matériau, apparaissent plusieurs bandes dont deux ont un intérêt particulier : l'une entièrement vide à 0 K appelée bande de conduction, et séparée d'une autre, appelée bande de valence, entièrement remplie à 0 K par un écart énergétique égal à 3,2 eV. Les rayonnements 5 responsables de la photocatalyse provoquent le passage d'un électron de la bande de valence vers la bande de conduction créant ainsi un trou noté h+ dans la bande de valence. 3.5 Quelle longueur d'onde maximum peut provoquer l'apparition d'une paire électron-trou ? 3.6 L'état excité obtenu peut être stabilisé par piégeage de l'électron. Dans le cas contraire, quel phénomène est observé ? 4. Cinétique de dégradation Chaque année, plus de 700 000 tonnes de colorants synthétiques sont produites dans le monde ; on estime qu'environ 10 % de ces colorants sont rejetés dans l'environnement. La plupart de ces composés sont des dérivés aromatiques difficilement biodégradables. Comme les méthodes existantes de purification des eaux industrielles sont extrêmement coûteuses, la photocatalyse, utilisant le rayonnement solaire et un matériau bon marché comme le TiO2 , laisse entrevoir une méthode alternative prometteuse dans le traitement des déchets. Dans la suite, on étudie la cinétique de minéralisation par photocatalyse d'un colorant synthétique utilisé dans l'industrie textile, l'acide bleu 80 (noté AB80, figure 3) dont le spectre d'absorption UV-visible en solution aqueuse est présenté figure 4. Figure 3 : Structure de l'acide bleu 80 Figure 4 : Spectre d'absorption UV-Visible de AB80 en solution aqueuse Afin d'étudier la cinétique de cette réaction de dégradation, trois expériences sont menées en parallèle. Une solution aqueuse du colorant AB80, tamponnée à pH = 6, est soumise à une aération prolongée. Un premier échantillon est placé dans le noir après y avoir ajouté TiO2 en poudre. Deux autres échantillons de cette solution sont exposés à la lumière solaire ; dans l'un des deux, TiO2 en poudre est ajouté. Des spectres d'absorption UV-visible sont enregistrés toutes les 10 minutes pour les trois échantillons afin de suivre la cinétique de la réaction (figure 5). 6 c /c0 Échantillon avec ajout de TiO2 non exposé à la lumière. Échantillon sans ajout de TiO2 exposé à la lumière solaire. N Échantillon avec ajout de TiO2 exposé à la lumière solaire. temps irradiation / min. Figure 5 : Évolutions du rapport (C/C0 ) en fonction du temps d'irradiation et des conditions (avec C : concentration en AB80 et C0 : concentration initiale en AB80) 4.1 Énoncer la loi de Beer-Lambert et les conditions de validité de cette loi. 4.2 En supposant les produits de dégradation incolores, montrer que la spectrophotométrie UV-visible permet de suivre la cinétique de cette réaction. 4.3 Quelle est la longueur d'onde optimale pour réaliser le suivi de la réaction ? 4.4 Que pouvez-vous conclure de l'analyse des courbes de la figure 5. On s'intéresse à l'échantillon ayant subi une irradiation en présence de TiO2 . La courbe donnant l'évolution du rapport (C/C0 ) (figure 5 (N)) est tracée en échelle logarithmique figure 6. 2.5 2 In(c0/c) 1.5 C0 C fonction du temps d'irradiation 1 Figure 6 : Évolution de ln 0.5 Å ã en 0 0 10 20 30 40 50 60 70 temps irradiation / min. On se propose d'étudier un mécanisme possible basé sur une première étape d'adsorption de AB80 en monocouche à la surface de TiO2 (première étape renversable) conduisant à la formation 7 de AB80 adsorbé (noté AB80ads ) suivie de l'oxydation par HO· (on se limitera à une seule espèce oxydante par souci de simplification) : Les cinétiques d'adsorption et de désorption, ainsi que la réaction d'oxydation, sont supposées d'ordre 1 par rapport aux réactifs. La concentration en radicaux HO· est supposée constante. 4.5 La cinétique d'adsorption peut être décrite par un modèle analogue à celui de la question 2.1, la concentration C en colorant remplaçant la pression P du diazote. Donner l'expression de la vitesse de la réaction d'oxydation du colorant en fonction de C, C (1 mol · L-1 ), k et K (constante d'adsorption), et [HO· ]. 4.6 Quelle hypothèse convient-il de faire pour que l'équation de vitesse décrivant ce mécanisme soit en accord avec les observations expérimentales ? En déduire la valeur de la constante de vitesse apparente en min-1 . 4.7 Au cours de ces essais, la concentration en AB80 est de l'ordre de 10-5 mol · L-1 et la constante d'adsorption K de ce même colorant sur la poudre de TiO2 est voisine de 5 · 103 . L'hypothèse précédente était-elle justifiée ? 8 Deuxième problème Utilisation de l'acide de Mosher en synthèse organique Ce problème traite de l'utilisation de l'acide de Mosher en chimie organique et présente deux parties indépendantes. Les molécules doivent être représentées complètement en tenant compte de leur stéréochimie. Des données spectroscopiques générales sont fournies en annexe à la fin de l'énoncé. 1. Acide de MOSHER et détermination de configurations absolues 1.1 Préparation de l'acide de MOSHER énantiomériquement pur L'acide de Mosher 1 (acide 3,3,3-trifluoro-2-méthoxy-2-phénylpropanoïque) est préparé de la façon suivante : O NaCN 3 Ph H2SO4 CH3-I 4 CF3 H2O, 2 2,2,2-trifluoroacétophénone O F3C Ph OH OMe 1 Acide de Mosher 1.1.1 Donner la structure de l'intermédiaire 3 (non isolé) et proposer un mécanisme pour expliquer sa formation. 1.1.2 Donner la structure de 4. 1.1.3 L'acide de Mosher 1 ainsi formé possède-t-il une activité optique ? Justifier la réponse. Ce composé 1 est dissous dans l'éthanol à température ambiante. La (R)-(+)-1-phényléthylamine : H3C H Ph NH2 est ajoutée à la solution précédente qui est chauffée puis abandonnée à température ambiante pendant 48 h. Le solide blanc 5 qui a précipité est récupéré (et le filtrat réservé), lavé et purifié deux fois par recristallisation. Les cristaux incolores obtenus sont dissous dans l'acide chlorhydrique dilué. Après 30 min d'agitation, le milieu est extrait avec de l'éther diéthylique, la phase éthérée est séchée puis le solvant évaporé sous pression réduite. Le liquide incolore obtenu est identifié par les données physiques comme étant l'acide 1 ; son pouvoir rotatoire spécifique à 25 C vaut []D = +65 deg · dm-1 · g-1 · mL. Par ailleurs, le filtrat précédent contenant le composé 6 est acidifié (HCl aqueux). Le mélange est traité comme précédemment et, après purification, un liquide incolore également identifié comme étant l'acide 1 est obtenu. La détermination de son pouvoir rotatoire spécifique à 25 C donne []D = -65 deg · dm-1 · g-1 · mL, et son atome de carbone stéréogène est de descripteur stéréochimique (ou configuration absolue) S. 1.1.4 Donner l'ordre de grandeur des constantes d'acidité respectives de 1 et de la 1-phényléthylamine. 9 1.1.5 En déduire les structures de 5 et 6. Quelle est la relation d'isomérie entre ces deux composés ? 1.1.6 Nommer la séquence permettant d'obtenir 5 et 6 purs à partir de l'acide 1. 1.1.7 En représentation de Cram, dessiner l'isomère dextrogyre de l'acide 1. 1.2 Propriétés spectroscopiques des dérivés de l'acide de MOSHER 1 L'acide de Mosher 1 et ses dérivés sont utilisés pour déterminer la configuration absolue des atomes de carbone stéréogènes des alcools et des amines par spectroscopie RMN 1 H. Cette technique repose sur la différence de déplacements chimiques entre deux isomères dérivés de l'acide 1, comme illustré ci-après. Le (-)-menthol est engagé dans la séquence réactionnelle suivante pour aboutir à la formation de 8 : 6 SOCl2 7 , 50 h OH H3C 8 Et3N, 2 h 9 CH3 10 (-)-menthol (R)-(+)-1 1 5 2 7 4 3 CH3 8 (-)-menthol Le composé 9 est préparé selon une séquence analogue à partir du (S)-(-)-1 et du (-)-menthol. 1.2.1 Déterminer les descripteurs stéréochimiques des centres stéréogènes du (-)-menthol. 1.2.2 Donner la formule semi-développée de 7 formé intermédiairement. 1.2.3 Donner la formule semi-développée de 8 en précisant le rôle de la triéthylamine. 1.2.4 Donner la formule semi-développée de 9. Quelle est la relation de stéréoisomérie entre 8 et 9 ? Les spectres RMN 1 H à 500 MHz de 8 et 9 sont enregistrés dans le chloroforme deutérié, puis analysés. Entre les deux spectres, les différences de déplacement chimique pour un atome Hi sont notées de la façon suivante (le cas échéant, si plusieurs atomes d'hydrogène sont portés par le même atome de carbone Ci , plusieurs valeurs sont données) : SR Hi = S (Hi ) - R (Hi ) avec S le déplacement chimique de l'atome Hi dans 9 (à partir de (S)-(-)-1). R le déplacement chimique de l'atome Hi dans 8 (à partir de (R)-(+)-1). L'analyse des spectres RMN 1 H des composés 8 et 9 a donné les résultats partiels portés dans le tableau 2 (la numérotation des atomes du (-)-menthol est celle définie plus haut) : Hi SR (Hi ) / Hz H2 -20 H3 -10 -15 H4 0 -15 H6 +30 +65 H7 -155 H8 -50 H9 -60 H10 +15 Tableau 2 : Différences de déplacements chimiques des protons Hi des composés 8 et 9 10 Des études conformationnelles montrent que les conformations de plus basse énergie de 8 et 9 sont celles dans lesquelles l'atome de carbone du groupement CF3 , le groupe CO2 et la liaison C1 -H du menthol se situent dans un même plan et sont dans les positions relatives dessinées cidessous (les autres substituants ne sont pas représentés ici). Pour la suite, les composés étudiés sont assimilés à une représentation simplifiée (dessin de droite) « omettant » le groupe CO2 (l'ordre de priorité déterminé au moyen des règles de Cahn, Ingold et Prelog est conservé). O F3C H H F3C O 1 1 1.2.5.a Dessiner le conformère le plus stable du (-)-menthol. 1.2.5.b En utilisant la représentation simplifiée introduite ci-dessus, dessiner, en projection de Newman (C1 devant), les conformères les plus stables des composés 8 et 9. Le signe de la différence de déplacements chimiques peut être interprété en terme de blindage/déblindage dû à la présence du groupe phényle. En effet, ce groupe se situe quasiment dans le plan orthogonal au plan contenant l'enchaînement CF3 -C-C1 -H défini précédemment. On rappelle que la présence d'un noyau aromatique partitionne l'espace en deux zones : a) une zone de blindage notée (+) dans laquelle une diminution de la valeur habituelle des déplacements chimiques est observée ; b) une zone de déblindage notée (-) dans laquelle la valeur habituelle des déplacements chimiques augmente. Ces zones sont représentées Figure 7. (+) Zone (+) : zone de blindage (-) (-) Zone (-) : zone de déblindage (+) Figure 7 : Partitionnement de l'espace en présence du noyau aromatique 1.2.6 Certaines valeurs de SR (Hi ) sont presque nulles. Interpréter ce résultat. Commenter les valeurs multiples observées pour H3 et H6 . 1.2.7 En utilisant ces modèles, et via des considérations de blindage/déblindage, interpréter les signes des différences de déplacements chimiques relevées dans le tableau 2. 1.2.8 Quelle est, en ppm, la plus grande différence de déplacements chimiques relevée dans le tableau 2 ? Quelle indication peut-on en tirer sur la position du groupe isopropyle ? 11 1.2.9 Pourquoi ne peut-on pas utiliser l'acide (R)-3,3,3-trifluoro-2-phénylpropanoïque à la place de 1 dans l'étude précédente ? 1.2.10 La méthode de Mosher est encore largement utilisée aujourd'hui. Une équipe japonaise l'a récemment mise à profit afin de déterminer la configuration absolue du centre stéréogène C13 de l'amphidinolide T1, une molécule organique complexe extraite d'algues marines, dont la structure est présentée ci-après. OH O 12 9 H H3C 10 H 6 O 5 CH3 14 Amphidinolide T1 15 11 8 7 13 16 H CH3 4 3 2 1 18 CH3 (La configuration du centre stéréogène C13 n'est pas définie) O O L'amphidinolide T1 est transformé dans un premier temps en 10 (à partir de (R)-(+)-1) et 11 (à partir de (S)-(-)-1), suivant le même protocole que celui utilisé pour le (-)-menthol. Les spectres RMN 1 H à 600 MHz de ces deux composés sont enregistrés en solution dans le chloroforme deutérié et analysés. Quelques valeurs des différences de déplacements chimiques entre 10 et 11 sont indiquées dans le tableau 3 : Hi H10 +36 SR (Hi ) / Hz H11 +12 +6 H14 -30 H15 -30 -90 Tableau 3 : Différences de déplacements chimiques des protons Hi des composés 10 et 11 Conclure quant à la configuration absolue de l'atome de carbone C13 . 2 Synthèse totale de l'amphidinolide T1 Une synthèse totale de l'amphidinolide T1 a été réalisée par assemblage des trois synthons A, B et C selon le schéma présenté ci-après. OMOM OH O TBSO CH3 OTBS CH3 H H3C O H H CH3 CH3 O SO2Ph H H3C O B CH3 CH3 O H Cl O A C O O Remarque : la structure de l'amphidinolide T1 omet volontairement la configuration absolue de l'atome C13 examinée dans la partie précédente. Il n'est pas nécessaire de la connaître pour traiter la suite du problème. 12 2.1 Préparation du synthon A Le synthon A est préparé en plusieurs étapes selon la séquence réactionnelle ci-après : O H3C OCH3 MgBr DIBAl-H 13 14 TsCl : H3C 16 puis H3O+ CH3 DIBAl-H : SO2Cl KCN 15 puis H3O+ CH2Cl2 12 HO TsCl, Et3N H3C H Al CH3 CH3 2.1.1 Donner la structure de 13 et proposer un mécanisme pour expliquer sa formation. 2.1.2 Le spectre IR de 14 fait apparaître une bande d'absorption intense vers 1740 cm-1 , et son spectre RMN 1 H présente un signal singulet vers 10 ppm. Donner sa structure. 2.1.3 Détailler le mécanisme de formation de 15, dont l'atome de carbone stéréogène nouvellement créé est de descripteur stéréochimique S. Est-ce le seul produit attendu dans cette transformation ? Si non, préciser la structure du (ou des) produit(s) également formé(s). 2.1.4 Donner la formule semi-développée de 16. 2.1.5 Le groupe fonctionnel introduit dans 16 est réduit en aldéhyde, pour conduire après traitement en milieu acide à une structure cyclique 17. Ce dernier est transformé en A en une étape non abordée ici. Proposer une structure pour 17 et un mécanisme permettant d'expliquer la cyclisation qui se produit en milieu acide. Nommer le groupe fonctionnel ainsi formé. 2.2 Préparation du synthon B La mise en place des différents centres stéréogènes du synthon B, dont la préparation est donnée ci-après, est contrôlée grâce à une réaction d'aldolisation diastéréosélective et énantiosélective. Dans ce qui suit, seule la diastéréosélectivité de cette réaction est étudiée. O CH3 Xp 18 1. Bu2BOTf Et3N 2. CH3 O O OH 1. LiBH4, Et2O CH3 Xp puis H3O+ 1. NaH, THF 20 19 2. MOMCl CH3 O Xp : O Ph 21 2. TBSCl, Et3N CH2Cl2 3. O3, CH2Cl2 puis Me2S N CH3 Bu2BOTf : Bu B O S CF3 Bu O O MOMCl : CH3OCH2Cl TBSCl : tBu(CH3)2SiCl THF : tétrahydrofurane 2.2.1 Le composé 18 est traité par un équivalent d'acide de Lewis, le trifluorométhanesulfonate de dibutylbore (Bu2 BOTf), et de triéthylamine dans le dichlorométhane à basse température (-78 C). 13 2.2.1.a Justifier le caractère acide de Lewis de Bu2 BOTf. 2.2.1.b Donner la structure des deux énolates susceptibles de se former lorsque 18 est traité par de la triéthylamine. 2.2.1.c Préciser la relation d'isomérie existant entre ces deux énolates. 2.2.2 Proposer un mécanisme simplifié pour expliquer la formation de 19. 2.2.3 La réaction d'addition de l'énolate sur la méthacroléine (CH2 =C(CH3 )CHO) implique très probablement un état de transition cyclique à 6 atomes en conformation chaise, dont un des sommets est occupé par l'atome de bore complexé par deux atomes d'oxygène : celui de la méthacroléine et celui de l'énolate. L'approche des réactifs préfigurant l'état de transition cyclique est schématisée ci-dessous : O B O 2.2.3.a Dessiner l'état de transition en conformation chaise correspondant à cette approche et placer seulement les substituants de la méthacroléine. Justifier la représentation ainsi choisie. 2.2.3.b Pour chacun des énolates formés (cf. question 2.2.1.b), dessiner l'état de transition cyclique de plus basse énergie en ajoutant au dessin précédent les substituants de l'énolate. Préciser dans chacun des cas le produit résultant d'un tel état de transition. 2.2.3.c L'analyse de 19 isolé montre que les groupements CH3 et OH sont en position relative syn. Conclure quant à la géométrie de l'énolate formé au cours de l'étape de déprotonation. 2.2.4 Préciser la structure de 20 et proposer un mécanisme pour expliquer la transformation de 19 en 20. 2.2.5 Sachant que l'action de LiBH4 sur 20 conduit à un composé présentant une bande d'absorption IR large vers 3600 cm-1 , donner la structure de 21. 2.2.6 Proposer des conditions expérimentales permettant d'obtenir B à partir de 21. 2.3 Préparation du synthon C Le synthon C est obtenu comme suit : Pr t-BuO O1 X'p 22 CH3 1. LDA, THF -78 °C 2. Br LiOH, THF 23 O 24 H2O EDCI, CH2Cl2 O2 X'p : O Ph N OH H3C EDCI : CH3 14 N CH3 1. CF3CO2H, CH2Cl2 25 2. (COCl)2, CH2Cl2 N C NEt Synthon C 2.3.1 Le composé 22 est d'abord traité par du diisopropylamidure de lithium (LDA) dans le tétrahydrofurane à basse température puis par du bromure d'allyle pour former 23. 2.3.1.a Justifier le choix du LDA en donnant un ordre de grandeur des pKA des différents couples acide-base mis en jeu. 2.3.1.b Dans les conditions opératoires utilisées, le cation lithium se complexe fortement avec les atomes d'oxygène numérotés O1 et O2 de la molécule 22. Montrer qu'il existe alors une approche privilégiée du bromure d'allyle. 2.3.1.c En tenant compte de la structure du synthon C, dessiner 23. 2.3.1.d En déduire la géométrie de l'intermédiaire majoritairement formé lors de la réaction entre 22 et le LDA. 2.3.2 Le spectre RMN 1 H de 24 présente à 11,3 ppm un pic élargi intégrant pour un proton, et son spectre IR présente deux bandes d'absorption à 3081 et 1708 cm-1 . Donner la formule semi-développée de 24. 2.3.3 La formation de 25 est accompagnée de celle d'un dérivé d'urée (contenant le motif NHC(=O)NH) ; en déduire la nature de la réaction qui s'est produite et la structure de 25. 2.4 Fin de la synthèse La dernière partie de la synthèse, comme décrite ci-dessous à partir de 26, implique une réaction de métathèse cyclisante qui permet de former 27, isolé sous la forme de deux stéréoisomères non séparés 27a et 27b dans un rapport 6 : 1. OMOM CH3 TBSO O H2, Pd/C métathèse O H H3C 27 H O 29 CH3CO2Et CH3 CH3 H Ph3P=CH2 28 26 O La réaction de métathèse permet de recombiner deux alcènes suivant le schéma : R1 R3 métathèse + R2 R4 R1 R3 R2 R4 + C2H4 2.4.1 Quelle est la relation de stéréoisomérie entre 27a et 27b ? Proposer une méthode analytique permettant de les différencier. 2.4.2 Donner la structure de 28. Pour quelle raison n'est-il pas nécessaire de séparer 27a et 27b ? 2.4.3 Nommer la transformation de 28 en 29 et donner la structure de 29. 2.4.4 Proposer une séquence de réactions et des conditions opératoires permettant d'obtenir l'amphidinolide T1 à partir de 29. 15 Tables spectroscopiques Déplacements chimiques en RMN 1 H -CH2 CH3-C=C CH3-Ar CH3-CO H-C=C H-Aromatique H2N-R -CHO HO-R (alcool) HO-Ar (phénol) HO-COR (acide) 0 2 4 6 8 10 (ppm) Bandes d'absorption infrarouge Type de liaison OH alcool OH acide carboxylique C=O ester C=O aldéhyde et cétone vibration valence valence valence valence nombre d'onde (cm-1 ) 3200-3670 2500-3200 1700-1745 1650-1740 intensité forte (large) forte à moyenne (large) forte forte abaissement de 20 à 30 cm-1 si conjugaison C=O acide carboxylique C=C valence valence 1680-1710 1625-1685 16 forte moyenne