
X/ENS Chimie PC 2015
| Thème de l'épreuve | Relations quantitatives entre structures moléculaires et propriétés physico-chimiques. Biosynthèse et synthèse totale des alcaloïdes. |
| Principaux outils utilisés | thermodynamique, cinétique chimique, oxydoréduction, chimie organique |
| Mots clefs | Hammett, alcaloïde, allosédamine, fawcettimine |
Corrigé
:👈 gratuite pour tous les corrigés si tu crées un compte
👈 l'accès aux indications de tous les corrigés ne coûte que 1 € ⬅ clique ici
👈 gratuite pour tous les corrigés si tu crées un compte
- - - - - - - - - - - - - -
👈 gratuite pour ce corrigé si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Énoncé complet
(télécharger le PDF)













Rapport du jury
(télécharger le PDF)








Énoncé obtenu par reconnaissance optique des caractères
ÉCOLE POLYTECHNIQUE ÉCOLES NORMALES SUPÉRIEURES
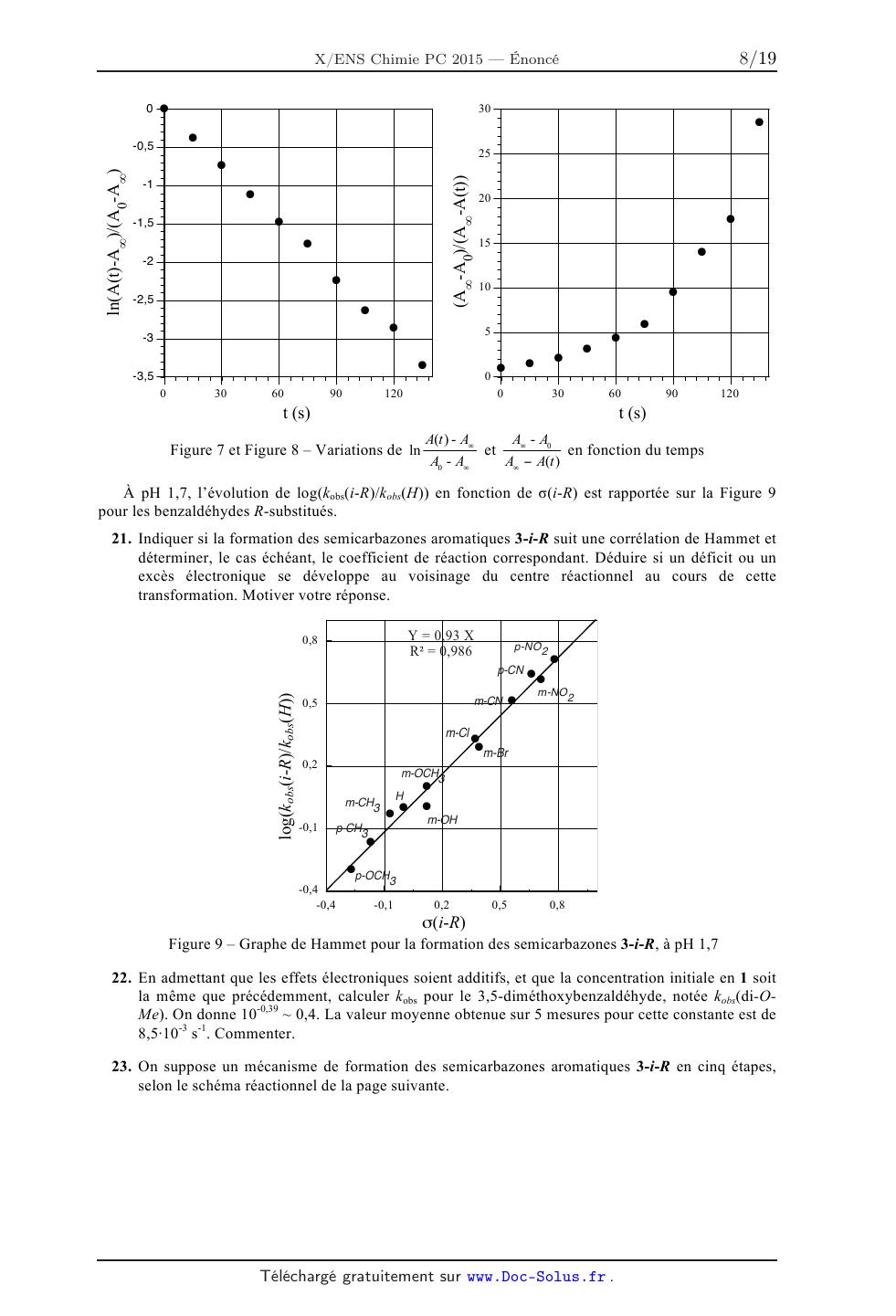
ÉCOLE SUPÉRIEURE DE PHYSIQUE ET DE CHIMIE INDUSTRIELLES
CONCOURS D'ADMISSION 2015
FILIÈRE PC
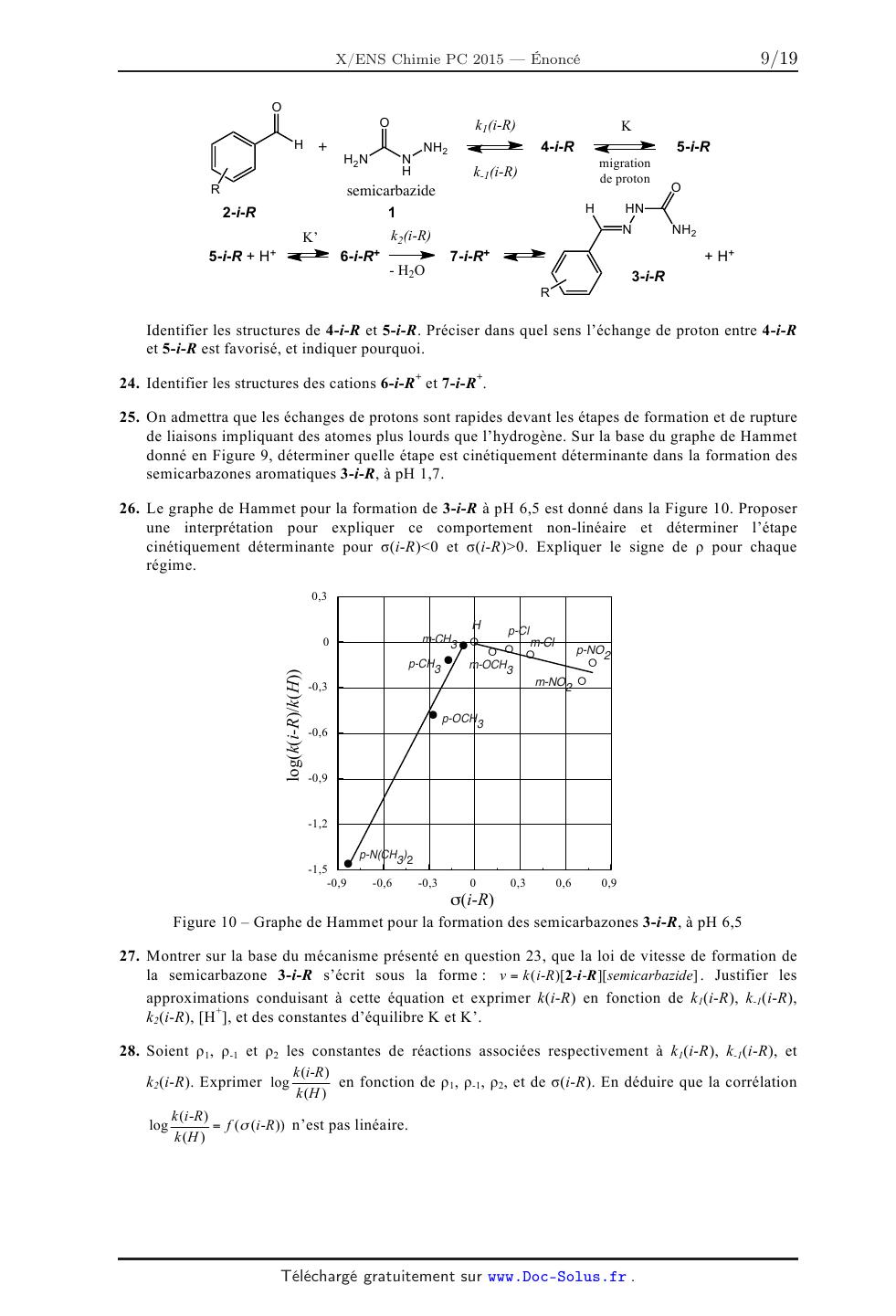
COMPOSITION DE CHIMIE A (XEULC)
(Durée : 4 heures)
L'utilisation des calculatrices n'est pas autorisée pour cette épreuve.
! !!
Premier problème
Les relations quantitatives entre structures moléculaires
et propriétés physico-chimiques
Prédire l'influence de substituants sur les propriétés physico-chimiques et la
réactivité d'une
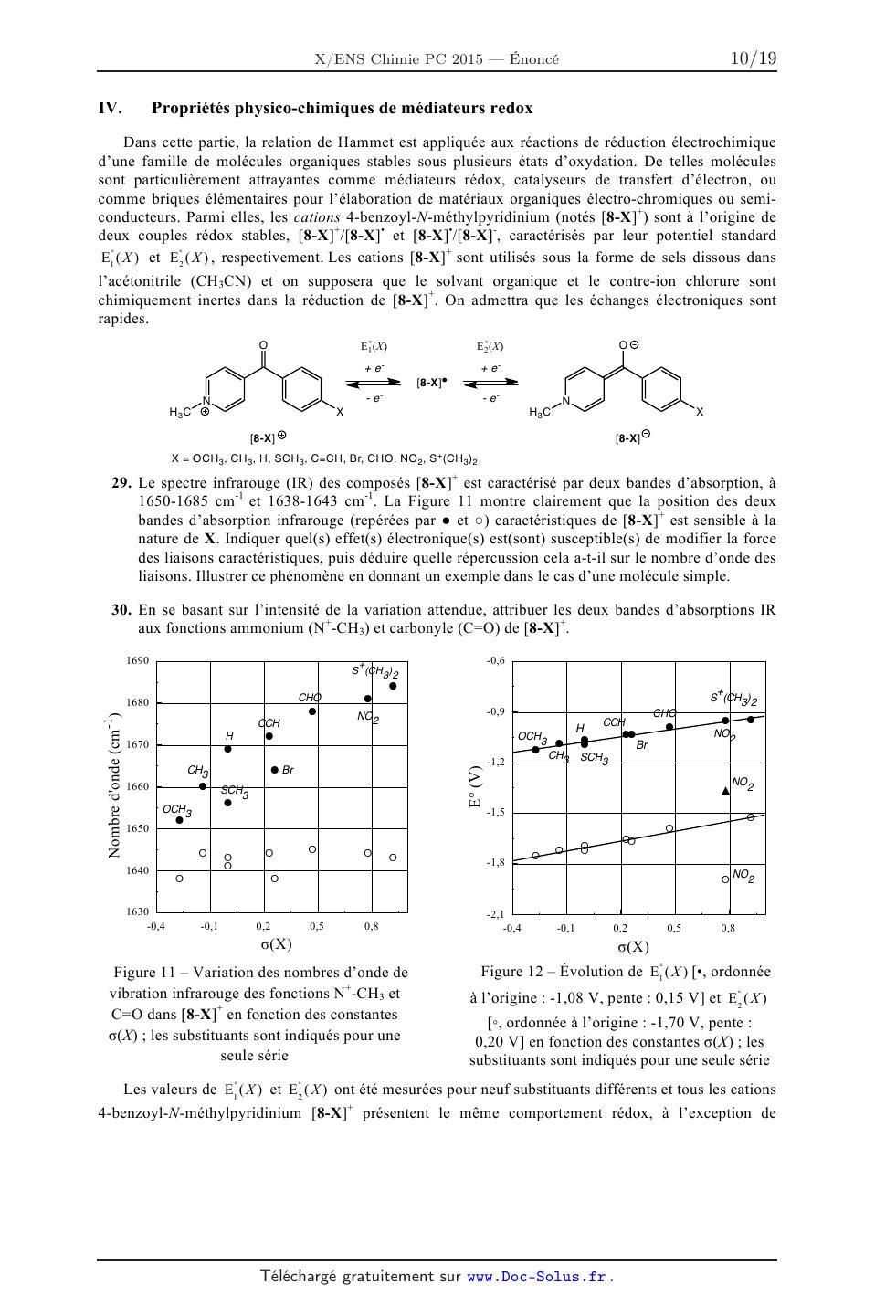
molécule organique a motivé, dès le début du XXème siècle, les efforts de
physico-chimistes. En 1940,
Louis Plack Hammet un professeur américain de l'Université de Columbia, a
publié un livre intitulé
« Chimie Organique Physique » qui a initié les bases d'une nouvelle discipline
pour de nombreux
chimistes organiciens. Il y a notamment proposé une équation empirique -
l'équation de Hammet - qui
permet de modéliser les variations de constantes d'équilibre ou de vitesse de
réactions mettant en jeu
des composés aromatiques en fonction de la nature des substituants qu'ils
portent. Cette méthode
prédictive a été appliquée avec succès à de nombreuses réactions organiques
pour essayer d'en
comprendre le mécanisme. Les constantes utilisées dans ces équations, bien que
critiquées pour leur
caractère empirique, ont très souvent permis la prédiction de constantes
d'équilibre et de vitesse de
réactions en solution très variées. Nous nous proposons dans ce problème
d'explorer le champ
d'applications de la relation de Hammet à plusieurs études thermodynamiques et
cinétiques en abordant
les trois problématiques suivantes :
·
·
·
équilibres acido-basiques en solution ;
mécanismes réactionnels ;
propriétés physico-chimiques de médiateurs rédox.
Les constantes thermodynamiques et cinétiques de ce problème sont des données
mesurées à 25 °C.
On prendra ln(10)RT/nF=0,06 V à 298 K.
I.
Constantes de Hammet et acidité des acides benzoïques substitués dans l'eau
La chimie organique physique initiée par Louis Hammet aborda notamment les deux
questions
suivantes :
-
quels effets peut avoir un groupe électro-donneur ou électro-attracteur, noté
R, sur l'énergie de
l'état de transition ou la stabilité de l'intermédiaire réactionnel formé au
cours d'une
transformation chimique ?
1
- Est-il possible de quantifier simplement le caractère donneur ou attracteur
d'un tel groupe pour
prédire l'amplitude de l'effet de ce groupe sur le cours d'une réaction ?
Pour tenter de répondre à ces questions, Hammet étudia notamment trois familles
de molécules
d'architectures voisines, celles des dérivés d'acides benzoïques substitués par
R, respectivement en
ortho (o), méta (m), et para (p) sur le cycle aromatique. L'étude consistait à
déterminer pour un type de
réaction, par exemple l'hydrolyse des benzoates d'éthyle R-substitués, les
constantes
thermodynamiques et/ou cinétiques de la réaction pour des molécules de chaque
famille (0, m, p), et à
relier ces résultats aux valeurs des pKa des acides benzoïques correspondants
R-substitués, i.e. les acides
carboxyliques portant le même groupement R substitué sur la même position. La
Figure 1 est l'une des
représentations imaginée par Hammet pour visualiser le comportement des
différentes familles i (i = 0
[A], i = m [0], et i = p [I]) suite à l'étude de l'hydrolyse en milieu éthanol
aqueux basique des
benzoates d'éthyle substitués par différents groupements R.
A
. p-N02 . esters para-substitués
. m-N02 . esters méta-substitués
A esters orlh0-substitués
yQ . m_Cl O'NO2 A 0
ou 0rth0
_9 ' p-Cl A o-F
. F méta / OH
p-
R/
. m-OCH
H . 3 para \
- A -
. m CH3 0 Cl acide benzoïque substitué
. p'OCH3
A o-CH3
>
log Ka
Figure 1 -- Graphe de Hammet pour l'hydrolyse des benzoates d'éthyle substitués
par des
groupements électro-actifs ; k est la constante de vitesse de la réaction
d'hydrolyse de l'ester éthylique
et Ka la constante d'équilibre d'acido-basicité du couple acide R-benzoïque/ion
R-benzoate
De ces résultats empiriques montrant les influences de la nature de la famille
i et de celle du
substituant R sur la réactivité des benzoates éthyliques étudiés, Hammet
détermina une relation entre
l'acidité des acides benzoïques R-substitués, la nature de la famille i, et la
nature du substituant R.
L'effet du substituant R sur les propriétés acides au sein des familles et sur
la vitesse d'hydrolyse des
esters correspondants est abordée dans cette première partie.
La relation proposée par Hammet le conduisit à définir une nouvelle constante
notée o(i--R) selon
l'équation (l) :
=U(i-R) ...
où K,,(H) et Ka(i--R) y sont respectivement les constantes d'acido-basicité
mesurées dans l'eau des
couples acide benzoïque/benzoate et acide benzoïque substitué/benzoate
substitué, en position méta (i =
m) ou para (i = p) par le groupement R. Dans un premier temps, on se focalisera
ainsi uniquement sur
ces deux familles. Le terme o(i--R) est appelée constante de Hammet et dépend
de i et de R.
1. Donner la définition des effets inductif (donneur noté +], attracteur noté
-1) et mésomère (donneur
noté +M, attracteur noté -M) exercés par un atome ou un groupe d'atomes, en
donnant les
caractéristiques générales de ces effets. lllustrer ces notions dans le cas des
acides benzoïques, en
précisant les effets I et M de trois substituants R possédant des effets de
natures différentes, et en
justifiant le propos par des représentations simples des délocalisations
électroniques.
2. Préciser les conditions expérimentales de l'hydrolyse d'un ester utilisées
pour ce travail, et en
déduire les précautions prises par Hammet afin de réaliser l'étude comparative
de la Figure 1.
Ecrire l'équation de la réaction d'hydrolyse du benzoate d'éthyle R-substitué
pour lequel i = p et
R=COCH3 (groupement acétyle) de constante de vitesse k(p-COCH3), puis
l'équation de la
réaction associée à la constante d'acidité Ka(p-COCH3). Prévoir dans quelle
partie du graphe se
trouve le point de valeurs [log Ka(p-COCH3), log k(p-COCH3)], grâce à l'étude
des effets
électroniques de R sur l'acidité, et à l'observation de la dispersion des
points sur la Figure 1. On
admettra que le groupe CO2 n'exerce aucun effet attracteur. Indiquer quel
substituant présenté
sur la Figure 1, il est préférable de choisir pour opérer une hydrolyse rapide
d'un ester de la
famille m.
3. Décrire quelles observations globales qualitatives peuvent être déduites des
résultats de la
Figure 1, en s'attachant à relever le comportement des familles et celui des
substituants.
Les constantes !(i-R) sont rassemblées dans le Tableau 1 qui contient aussi une
évaluation semiquantitative de l'intensité des effets électroniques des
substituants R suivant la nature inductive et/ou
mésomère de ces effets (échelle en +I, -I, +M et M).
R
NMe2
NH2
CO2 OMe
CH3 H
!(m-R)
0,15
0,16
0,10
0,12
0,07
!(p-R)
0,83
0,66
0,00
...
0,17
Effet I
-
-
+
-
Effet M
+++
++
0
++
Br
+
NO2
N+Me3
N2
0,38
0,71
0,88
1,76
0,45
0,50
0,78
0,82
1,91
-
--
--
---
----
----
+
--
---
--
...
----
F
Cl
I
CO2Et COMe
0
0,34
0,37
0,39 0,35
0,37
0
0,06
0,23
0,24 0,28
+
---
--
--
0
++
+
+
Tableau 1 Constantes !(i-R) en fonction du substituant R et de sa position
sur le cycle benzénique,
et effets électroniques inductifs et mésomères des substituants R. Les cases
renseignées « ... » sont
l'objet d'une question spécifique. Le groupement méthyle CH3 a été abrégé en Me
4. Indiquer une méthode expérimentale permettant de déterminer !(i-R). L'acide
p-méthoxybenzoïque (R = p-OMe) possède un pKa de 4,49, alors que celui de
l'acide benzoïque est de 4,22.
Déduire la constante !(p-OMe), commenter son signe et le comparer au signe de
!(m-OMe).
Grâce au modèle des effets électroniques, interpréter l'influence de la
position du substituant
OMe sur l'acidité des acides m- et p-méthoxybenzoïques.
5. Pour le substituant R=N+Me3, le rapport calculé
! (m-N + Me3 )
=1,07 est voisin de 1. Déduire une
! (p-N + Me3 )
propriété du substituant et proposer une interprétation.
6. Afin de séparer les effets électroniques gouvernant la valeur des grandeurs
!(i-R), un autre
chercheur, le professeur Robert W. Taft, a proposé d'exprimer les constantes
!(i-R) sous la forme
d'une somme pondérée de deux paramètres - notés !'(R) et !''(R) - dépendant
uniquement de la
nature du substituant R mais pas de sa position sur le cycle aromatique :
! (p-R)=! '(R)+! ''(R)
! (m-R)=! '(R)+"! ''(R) avec " <1 D'après les résultats des questions 4 et 5, déduire la signification respective des paramètres !'(R) et !''(R). Préciser le rôle du coefficient !. 3 7. Les grandeurs o(p-R) et o(m-R) du Tableau 1 peuvent être prédites semi-empiriquement pour l'ensemble des substituants R en prenant a = 1/z dans les équations ci-dessus et en utilisant les paramètres de Taft du Tableau 2. Dans le cas du substituant R : N+Me3, déterminer les valeurs de ces deux paramètres de Taft a partir du Tableau 1, et discuter de leur cohérence avec l'interprétation donnée en question 5. Calculer o'(R) et o"(R) pour la série des halogènes et commenter les tendances observées. 8. D'après le Tableau 1 et la Figure 1, choisir le substituant R 0'9 P+Me et sa position pour obtenir le dérivé d'acide benzoïque 3' présentant la plus forte acidité. Calculer sa constante o'(R). 06 . . , . ' (CHSJgV DCH3 SOME. Expliquer sa valeur par rapport a celle du substituant fluor. . CH {CONH . Parmi les substituants R : C2H5, CHO, SMe, et SO;Me, «2,350 ' dire lequel est caractérisé par les constantes o(m-R) : 0,60 0,3 N:2 soe° C'°"o°.CF et o(p-R) : 0,72, grâce aux calculs des paramètres de Taft. 3 06H5 °C CFA/"ECO . 9. Au regard de la modélisation étudiée, commenter la représentation de la Figure 2 ci-contre, en explicitant la signification des axes. Préciser si les différents domaines COO'_ , . . , . -0,3 delimites dans ce graphique sont en accord avec le _136 _132 _038 _034 0 034 Tableau 1. Figure 2 -- Effets des substituants Substituant R Structure o'(R) 6' '(R) Substituant R Structure o'(R) 6' '(R) Acétamido CH3CONH 0,42 -0,42 Éthynyle HCEC 0,19 0,04 Acétoxy CH3C02 0,29 0,16 Hydrogène H 0 0 Acétyle CH3CO 0,26 0,24 Hydroxy HO 0,61 -0,98 Amino NH2 0,34 -1 Méthoxy CH3O 0,51 -0,78 t-Butyle (CH3)3C 0 -0,2 Méthoxycarbonyle CH3OCO 0,29 0,16 Carboxy H02C 0,29 0,16 Méthyle CH3 0,03 -0,2 Cyano NEC 0,46 0,2 Méthylthio CH3S 0,3 -0,3 Diméthylamino (CH3)2N 0,51 -1,34 Nitro N02 0,64 0,14 Éthoxy C2H50 0,44 -0,68 Phényle C6H5 0,11 -0,1 Éthényle CH2=CH -0,16 0,2 Trifiuorométhyle CF3 0,32 0,22 Éthyle C2H5 0,01 -0,16 Triméthylsilyle (CH3)3 Si -0,01 -0,06 Tableau 2 -- Valeurs des paramètres de Taft G'(R) et o"(R) pour les substituants R I]. Corrélations de Hammet et équilibres acid0-basiques Dans cette partie, on s'intéresse à l'effet du substituant R sur les propriétés acides au sein des familles de phénols substitués en toute position (i = o, m, p). Cet effet s'illustre sur le graphe de la Figure 3 de la page suivante, où est représentée la relation entre le pKa'(i--R) des couples phénols/phénolates substitués par un groupement R en fonction de celui des couples acides benzoique/benzoates substitués en même position par le même groupement R. On se propose de tracer un graphe avec certains points de la Figure 3 en reportant respectivement pour chacun d'eux : en abscisse la valeur de la constante o(i--R) définie en partie I, en ordonnée la valeur Kü'(i-R) K, '(H ) substituant R et sa position sur le cycle benzénique. La droite de régression tracée pour l'ensemble des points a pour équation Y : 2,2X avec un coefficient de détermination R2 = 0,983. log . Sur ce graphe représenté en Figure 4 ont été indiquées pour chaque point la nature du . phénols m-- et p--R--substitués * phénols o-R-substitués OH pK,'(i--R> -- pK,'(H> ,@
1 \
0,5
p--CH
GCH3 * 177 H 63 . p--OMe
. o--OMe 3
pKa(l--R)--pKa(I--Dl | | * | | * 0 |
0 -1,5 1,2 -o,9 C -0,6 -o,3 p--F 0 ' 0,3
O' 6H5 m--OMe . m--NH2
R// | OH o--OH* p--CIO . _ 05
F m--OH
\ m-- . p--Br
m--Br .
.
0_F m'Cl - '1
*
0--C/ m--CN .
o--Br * _ _1 5
* m--N02 . '
-- -2
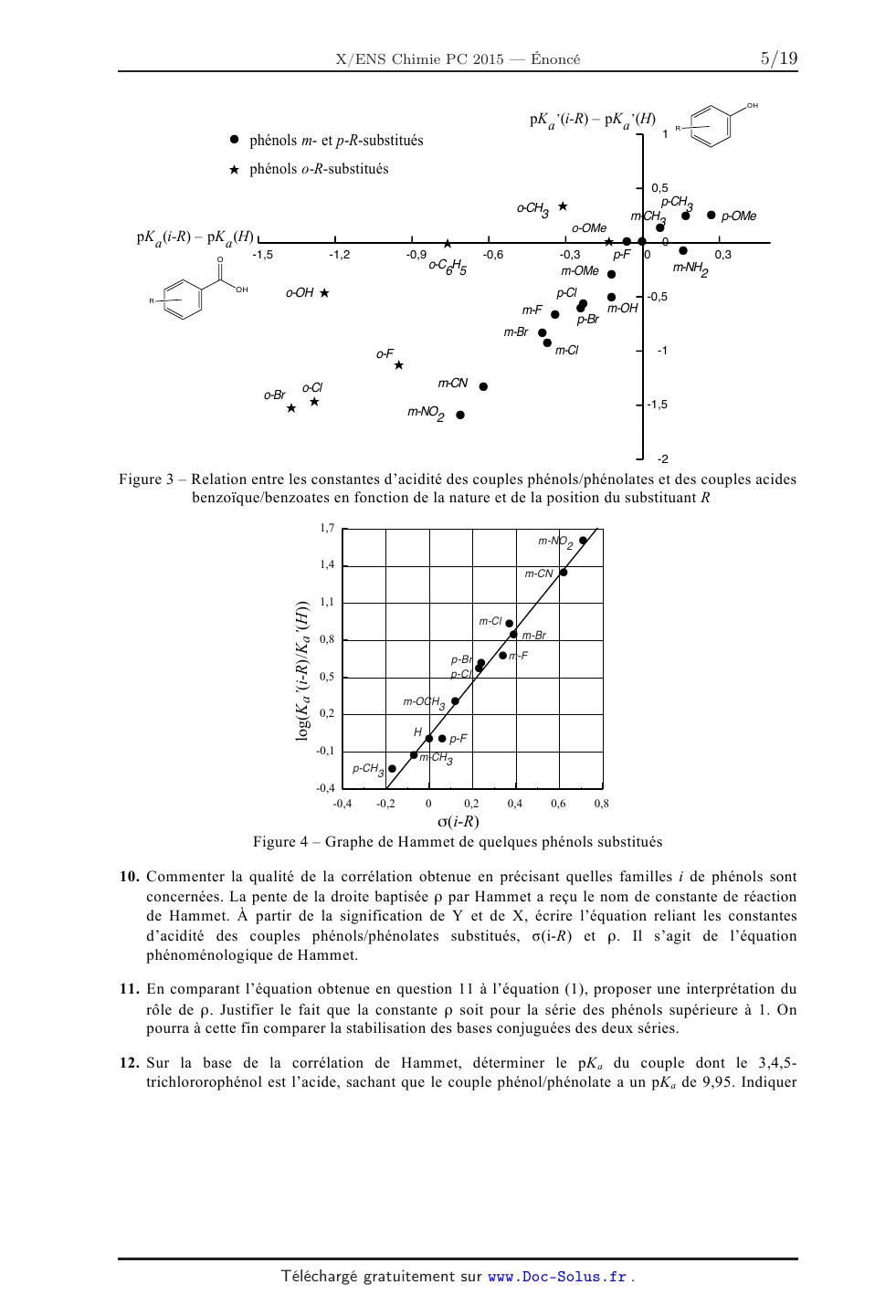
Figure 3 -- Relation entre les constantes d'acidité des couples
phénols/phénolates et des couples acides
benzoïque/benzoates en fonction de la nature et de la position du substituant R
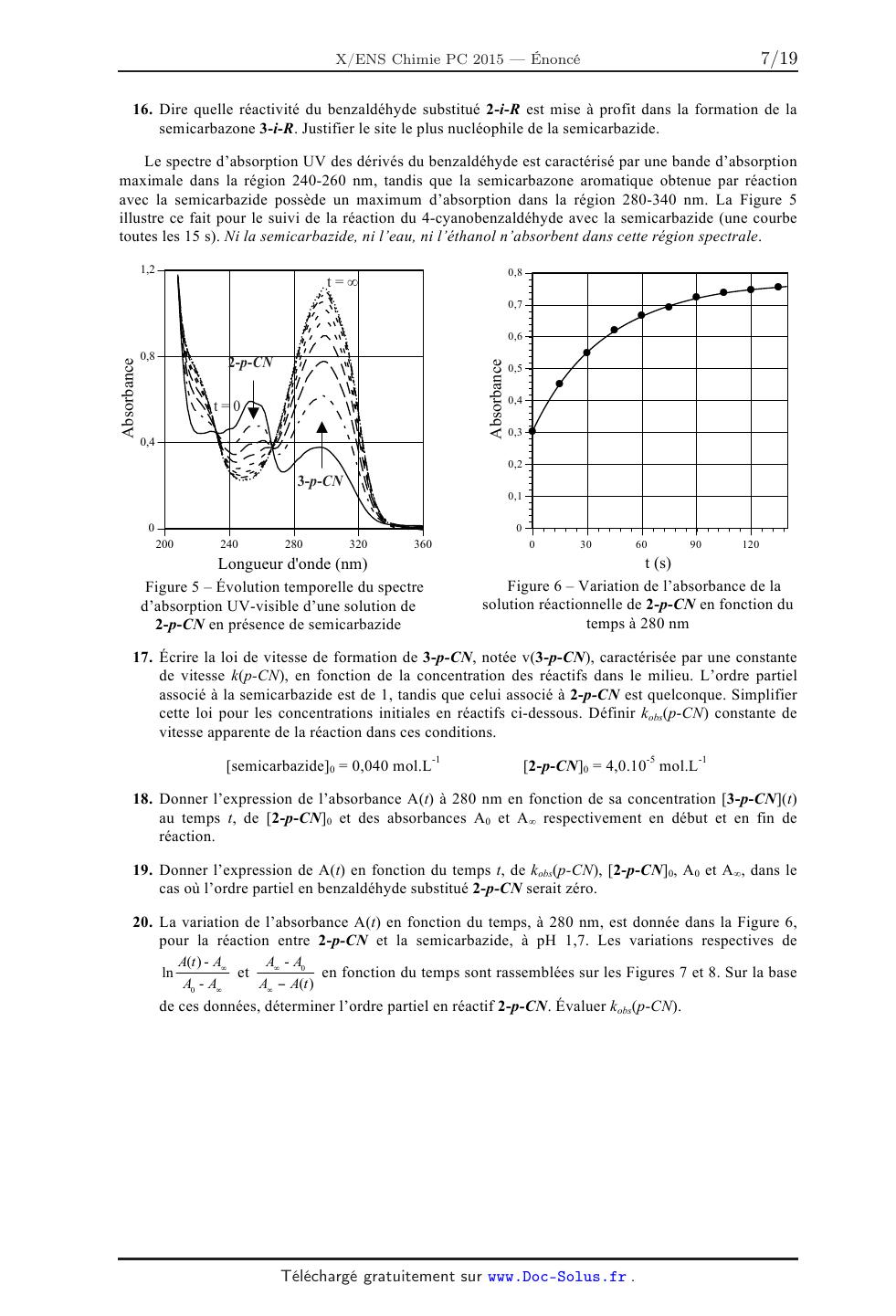
1,7
m-NOZ//
1,4
m--Cÿ'
EUR 1,1
m-Cl . /
V
' @ 0,8 /' m-Br
ä p-Br /m-F
Î 0,5 P-CI,
>/
@ m-OCH /
3
Ë3 0,2
--2 H . p-F
--O,1 m'CH3
p-CH3 .
-0,4 -
--0,4 --0,2 0 0,2 0,4 0,6 0,8
o(i--R)
Figure 4 -- Graphe de Hammet de quelques phénols substitués
10. Commenter la qualité de la corrélation obtenue en précisant quelles
familles i de phénols sont
11.
12.
concernées. La pente de la droite baptisée p par
Hammet a reçu le nom de constante de réaction
de Hammet. À partir de la signification de Y et de X, écrire l'équation reliant
les constantes
d'acidité des couples phénols/phénolates substitués, o(i-R) et p. Il s'agit de
l'équation
phénoménologique de Hammet.
En comparant l'équation obtenue en question 11 à l'équation (l), proposer une
interprétation du
rôle de p. Justifier le fait que la constante p soit pour la série des phénols
supérieure à 1. On
pourra à cette fin comparer la stabilisation des bases conjuguées des deux
séries.
Sur la base de la corrélation de Hammet,
déterminer le pKa du couple dont le 3,4,5-
trichlororophénol est l'acide, sachant que le couple phénol/phénolate a un pKa
de 9,95. Indiquer
quelle hypothèse doit être faite pour appliquer cette méthode. En réalité, le
couple 3,4,5trichlororophénol/3,4,5-trichlororophénolate présente un pKa de
7,75. Commenter.
13. D'après la Figure 3, une série de points ne respectent pas la corrélation
de Hammet. Reconnaître
la famille correspondante, puis dessiner une molécule de cette famille issue de
la Figure 3, en
précisant la(es) raison(s) expliquant un tel écart. Justifier la position de
son point représentatif sur
la Figure 3.
14. Les couples dont le 4-hydroxybenzoate d'éthyle (éthylparabène) et le
para-nitrophénol sont les
acides ont respectivement un pKa dans l'eau de 8,37 et 7,16. Indiquer comment
se placent les
points représentatifs de ces molécules sur le graphe de la Figure 4. Commenter
ce résultat, puis
fournir une explication à la position trouvée pour le point du para-nitrophénol
sur le graphe
précédent. Envisager une modification possible de l'équation de Hammet pour
solutionner cette
anomalie.
Le pKa associé à la déprotonation de dérivés du triphénylméthane (proton
indiqué) est donné cidessous, pour la molécule non-substituée (A) et ses
analogues substitués par un à trois groupements
nitro (B, C et D), dans le DMSO (CH3SOCH3). On admettra que la constante
associée à la réaction de
déprotonation de ces composés suit la corrélation de Hammet.
NO2
NO2
C
H
C
H
NO2
C
H
C
H
O2N
pKa =
O2N
NO2
A
B
C
D
30,8
16,8
14,4
12,7
15. Préciser pourquoi le pKa de ces composés est mesuré dans le DMSO. Ecrire
l'équation de
formation de la base conjuguée de B par réaction de B avec la base conjuguée du
DMSO. En
s'inspirant de la relation de Hammet, montrer que l'on peut expliquer
l'évolution du pKa des
couples dont les composés A à D sont les acides.
III.
Les corrélations de Hammet et les mécanismes réactionnels
La relation phénoménologique de Hammet, exprimée dans l'équation trouvée en
question 10 pour
des grandeurs purement thermodynamiques, peut être étendue aux constantes de
vitesse de réactions
mettant en jeu des réactifs aromatiques substitués comme entrevu dans la
première partie. L'équation de
Hammet relie alors les constantes de vitesse pour le réactif substitué (k(i-R))
et non-substitué (k(H)) par
une nouvelle constante $, caractéristique de la réaction mise en jeu, et la
constante !(i-R) définie
précédemment.
Dans cette partie, nous nous proposons d'étudier, grâce à la relation de Hammet
le mécanisme de
formation de semicarbazones aromatiques en milieu éthanol-eau, par réaction
entre un benzaldéhyde
substitué par le groupement R en position méta ou para (i = m ou i = p) et la
semicarbazide 1, selon le
bilan suivant :
O
O
H
O
H2N
N
H
NH2
N
H
+
HN
R
1
R
2-i-R
6
3-i-R
NH2
+
H2O
16. Dire quelle réactivité du benzaldéhyde substitué 2-i--R est mise à profit
dans la formation de la
semicarbazone 3-i--R. Justifier le site le plus nucléophile de la semicarbazide.
Le spectre d'absorption UV des dérivés du benzaldéhyde est caractérisé par une
bande d'absorption
maximale dans la région 240-260 nm, tandis que la semicarbazone aromatique
obtenue par réaction
avec la semicarba2ide possède un maximum d'absorption dans la région 280-340
nm. La Figure 5
illustre ce fait pour le suivi de la réaction du 4-cyanobenzaldéhyde avec la
semicarbazide (une courbe
toutes les 15 s). Ni la semicarbazide, ni ! 'eau, ni ! 'éthanol n 'absorbent
dans cette région spectrale.
L2 (L8
0,7 j
0»6 : /
033--
8 8 0,5
: = : /
c6 ce 2
@ "'à 0,4/
3 B ï
0,3
< 0,4 < 0,2 0,1 g 0 | 0 * | | | | I | I I I I I | I | I I | 200 240 280 320 360 0 30 60 90 120 Longueur d'onde (nm) t (5) Figure 5 -- Évolution temporelle du spectre Figure 6 -- Variation de l'absorbance de la d'absorption UV-visible d'une solution de solution réactionnelle de 2-p-CN en fonction du 2-p-CN en présence de semicarbazide temps à 280 nm 17. Écrire la loi de vitesse de formation de 3-p-CN, notée v(3-p-CN), caractérisée par une constante de vitesse k(p--CN), en fonction de la concentration des réactifs dans le milieu. L'ordre partiel associé à la semicarbazide est de 1, tandis que celui associé à 2-p-CN est quelconque. Simplifier cette loi pour les concentrations initiales en réactifs ci-dessous. Définir kobs(p-CN) constante de vitesse apparente de la réaction dans ces conditions. [semicarbazide]o = 0,040 mol.L'1 [2-p-CN]O = 4,010"5 mol.L'1 18. Donner l'expression de l'absorbance A(t) à 280 nm en fonction de sa concentration [3-p-CN](t) au temps t, de [2-p-CN]O et des absorbances A0 et A... respectivement en début et en fin de réaction. 19. Donner l'expression de A(t) en fonction du temps t, de k...(p--CN), [Lp-CN]... A0 et A..., dans le cas où l'ordre partiel en benzaldéhyde substitué 2-p-CN serait zéro. 20. La variation de l'absorbance A(t) en fonction du temps, à 280 nm, est donnée dans la Figure 6, pour la réaction entre 2-p-CN et la semicarbazide, à pH 1,7. Les variations respectives de A(t)-Aæ t ADO--A0 AO - Aoe " Aoe - A(t) de ces données, déterminer l'ordre partiel en réactif 2-p-CN. Évaluer k...(p-CN). ln en fonction du temps sont rassemblées sur les Figures 7 et 8. Sur la base 1n(A(t)-Aoe)/(AO-Aoe) À pH 1,7, l'évolution de log(kobs(i-R)/kobs(H)) en fonction de o(i-R) est rapportée sur la Figure 9 0 ? -0,5 : ° : o -1 : a : O. Expliquer le signe de p
pour chaque
régime.
0,3
H _
0 "' CH3 @ p Clin-Cl
. "\o\o\g\ p-N02
A p-CHS m-OCH3 \0
E -0 3 m'N02 0
Ë ,
> p-OCH
°Ï -0,6 3
&" /
V
CD
2 -0,9 /
-1,2 /
ÂN(CHÿ2
_1)5 . . . . .
-0,9 -0,6 -0,3 0 0,3 0,6 0,9
o(i-R)
Figure 10 -- Graphe de Hammet pour la formation des semicarbazones 3-i--R, à pH
6,5
Montrer sur la base du mécanisme présenté en question 23, que la loi de vitesse
de formation de
la semicarbazone 3-i--R s'écrit sous la forme : v =
k(i--R)[2-î-R][semicarbazz'de]. Justifier les
approximations conduisant à cette équation et exprimer k(i-R) en fonction de
k;(i-R), k.](i-R),
kg(i-R), [H+], et des constantes d'équilibre K et K'.
Soient pl, p-1 et p2 les constantes de réactions associées respectivement à
k;(i-R), k.](i-R), et
. . k(i-R)
k -R . E 1
2(z ) xpr1mer og k(H
en fonction de pl, ..., p2, et de o(i-R). En déduire que la corrélation
log% = f (GU-R)) n'est pas linéaire.
IV.
Propriétés physico-chimiques de médiateurs redox
Dans cette partie, la relation de Hammet est appliquée aux réactions de
réduction électrochimique
d'une famille de molécules organiques stables sous plusieurs états d'oxydation.
De telles molécules
sont particulièrement attrayantes comme médiateurs rédox, catalyseurs de
transfert d'électron, ou
comme briques élémentaires pour l'élaboration de matériaux organiques
électro-chromiques ou semiconducteurs. Parmi elles, les cations
4-benzoyl-N-méthylpyridinium (notés [8-X]+) sont à l'origine de
deux couples rédox stables, [8-X]+/[8-X]· et [8-X]·/[8-X]-, caractérisés par
leur potentiel standard
+
E1° ( X ) et E°2 ( X ) , respectivement. Les cations [8-X] sont utilisés sous
la forme de sels dissous dans
l'acétonitrile (CH3CN) et on supposera que le solvant organique et le
contre-ion chlorure sont
chimiquement inertes dans la réduction de [8-X]+. On admettra que les échanges
électroniques sont
rapides.
O
E1°(X)
E2°(X)
+ e-
+ e-
O
[8-X]
- e-
N
- e-
N
X
H3C
X
H3C
[8-X]
[8-X]
X = OCH3, CH3, H, SCH3, C!CH, Br, CHO, NO2,
S+(CH
3)2
+
29. Le spectre infrarouge (IR) des composés [8-X] est caractérisé par deux
bandes d'absorption, à
1650-1685 cm-1 et 1638-1643 cm-1. La Figure 11 montre clairement que la
position des deux
bandes d'absorption infrarouge (repérées par & et ') caractéristiques de [8-X]+
est sensible à la
nature de X. Indiquer quel(s) effet(s) électronique(s) est(sont) susceptible(s)
de modifier la force
des liaisons caractéristiques, puis déduire quelle répercussion cela a-t-il sur
le nombre d'onde des
liaisons. Illustrer ce phénomène en donnant un exemple dans le cas d'une
molécule simple.
30. En se basant sur l'intensité de la variation attendue, attribuer les deux
bandes d'absorptions IR
aux fonctions ammonium (N+-CH3) et carbonyle (C=O) de [8-X]+.
!")$
!'#)
"
! #$%&'(
!
!"#
!
!
!
!"'$
!
!"#$
!
!'#(
!"#
!!"
!!
! !"
!"#
!""$
!!"
!
"
! #$%&'(
!
+,-./0
-./012345.642378/*!9
!"($
!"#$
!
!$#"
!
!"#$
! !
! !" !
# !"#$
!"#
!$#&
"
"
"
!"#$
*$+%
!"
! !
!"#
#
!"&$
!"%$
!"#
!
"
"
"
"
"
"
"
!$#%
"
"
"
"
""
" !"#
"
*$+!
$+,
$+&
!"#$
!'#*
$+(
7:9
!'#$
'#"
'#&
'#%
.10
!
!
Figure 12 Évolution de E1° ( X ) [·, ordonnée
Figure 11 Variation des nombres d'onde de
vibration infrarouge des fonctions N+-CH3 et
C=O dans [8-X]+ en fonction des constantes
!(X) ; les substituants sont indiqués pour une
seule série
à l'origine : -1,08 V, pente : 0,15 V] et E°2 ( X )
[!, ordonnée à l'origine : -1,70 V, pente :
0,20 V] en fonction des constantes !(X) ; les
substituants sont indiqués pour une seule série
Les valeurs de E1° ( X ) et E°2 ( X ) ont été mesurées pour neuf substituants
différents et tous les cations
4-benzoyl-N-méthylpyridinium [8-X]+ présentent le même comportement rédox, à
l'exception de
10
[8-NO2]+ qui possède trois états de réduction stables et qui sera discuté dans
les questions de la fin du
problème. L'évolution de E1° ( X ) et E°2 ( X ) en fonction des constantes !(X)
est donnée dans la
Figure 12.
31. Écrire l'équation de Hammet pour ces deux réactions de réduction, puis en
déduire l'expression
de E1° ( X ) et E°2 ( X ) en fonction de !(X), E1° (H ) et E°2 (H ) , et de
deux nouvelles constantes de
réaction de Hammet notées $'1 et $'2 que l'on précisera. On pourra noter K1 et
K2 les constantes
d'équilibre des réactions de réduction mentionnées plus haut.
32. La réduction mono-électronique d'une fonction cétone conduit à
la formation d'un radical anion selon le schéma ci-contre. Écrire
la structure de Lewis du radical anion obtenu par réduction monoélectronique de
la benzophénone ((C6H5)2C=O). Montrer que ce
radical anion est stabilisé par résonance.
33. Le cation N-méthylpyridinium peut également subir une réduction
mono-électronique par
addition d'un électron sur la fonction iminium C=N+. Écrire la structure de
Lewis du radical
obtenu par réduction mono-électronique du cation N-méthylpyridinium, et montrer
que ce radical
est aussi stabilisé par résonance.
34. Montrer que la réduction mono-électronique de [8-X]+ peut conduire à deux
radicaux distincts
dont on explicitera les formules semi-développées. Sachant que les potentiels
standard de
réduction de la benzophénone ((C6H5)2C=O) et du cation N-méthylpyridinium sont
respectivement -1,78 V et -1,37 V, déterminer la structure de [8-X]·.
35. À partir des résultats de la Figure 12, calculer $'1 et $'2. Interpréter le
signe de $'1, $'2, et $'2-$'1.
36. D'après la Figure 12, le cation [8-NO2]+ peut subir trois étapes de
réduction mono-électroniques
dont les potentiels standards sont bien séparés. On note [8-NO2]·, [8-NO2]! et
[8-NO2]2! les trois
états de réduction de [8-NO2]+. Déterminer et justifier la structure de
[8-NO2]·.
37. Proposer une structure pour [8-NO2]! et [8-NO2]2!. On rappelle que le
potentiel standard du
couple [nitrobenzène]/[nitrobenzène]! vaut -1,54 V. Indiquer si l'anion
[8-NO2]! est radicalaire, et
proposer une explication.
38. Évaluer les potentiels standards d'oxydoréduction des couples
[8-NO2]·/[8-NO2]! et [8-NO2]!/
[8-NO2]2!. Dire pourquoi ils ne suivent pas la corrélation de Hammet de la
Figure 12.
39. Montrer que l'espèce [8-NO2]2! peut s'écrire sous la forme [8-Z]!, pour
laquelle la nature du
substituant Z sera précisée. Estimer !(Z) d'après les résultats de la
corrélation portant sur le
deuxième transfert électronique. Préciser votre démarche. Justifier le signe de
!(Z) et comparer sa
valeur à celle des constantes de Hammet données dans le Tableau 1.
40. En conclusion, expliciter en dix lignes au maximum la démarche de Hammet.
En quoi le modèle
est-il fécond et son application toujours d'actualité ?!
11
Deuxième problème
Alcaloïdes - Biosynthèse et synthèse totale
Les alcaloïdes sont des substances naturelles azotées basiques comportant
systématiquement, à
quelques exceptions près, un hétérocycle azoté. Métabolites secondaires
produits à partir d'aminoacides par des végétaux, des champignons ou certaines
espèces animales, les alcaloïdes constituent le
groupe de produits naturels le plus grand et le plus diversifié. Leurs
propriétés biologiques très variées
(analgésiques, antibiotiques, antifongiques, antitumorales, antipaludiques ...)
justifient l'intérêt qu'ils
suscitent. Il est intéressant de noter qu'à l'heure actuelle, 80% des principes
actifs des médicaments
contiennent un atome d'azote dans leur structure, et pour plus de la moitié
d'entre eux, il s'agit d'un
hétérocycle azoté. Dans ce problème, nous nous intéresserons à la famille des
alcaloïdes issus du
métabolisme de la lysine, en étudiant les étapes initiales de leur biosynthèse
ainsi que la synthèse totale
de deux composés de cette famille : l'allosédamine et la fawcettimine.
I.
De la lysine aux pipéridines substituées : étude de deux étapes de la
biosynthèse
La lysine [1] est un acide "-aminé naturel dont la transformation en
#1-pipéridéine [4], constitue le
point de départ de la biosynthèse de certains alcaloïdes. Les premières étapes
de cette transformation
sont habituellement représentées par le Schéma 1 suivant :
4
5
6
4
3
2
5
Oxydation
Décarboxylation
H2N H2N
H2N H2N
CO2H
H
[1]
H2N
[2]
H
O
6
[3]
3
1
N
[4]
2
1
Schéma 1 Formation de la # -pipéridéine [4] à partir de la lysine
La première étape de la biosynthèse de [4] est la décarboxylation de la
lysine [1] catalysée par une enzyme (décarboxylase) associée au phosphate
de pyridoxal [5] comme cofacteur. Ce cofacteur réagit tout d'abord avec la
lysine [1] pour conduire intermédiairement à une imine [6].
O
HO
H
OPO32
N
De manière générale, les aldéhydes réagissent avec les amines primaires Me
[5]
H
pour conduire à des produits de condensation de formule générale A, appelés
imines, et de l'eau (Schéma 2). La formation des imines A est un processus
équilibré renversable. Cet
aspect est particulièrement important dans les transformations biosynthétiques
étudiées.
O
N
+
R
R'
R'NH 2
+
H
R
H
H 2O
A
Schéma 2 Formation d'une imine à partir d'un aldéhyde
1. Proposer un mécanisme pour cette transformation du Schéma 2 en faisant
intervenir une catalyse
par un acide de Brønsted fort.
2. Pour synthétiser des imines en laboratoire avec de bons rendements, une
technique expérimentale,
dans le cas où les composants sont peu volatils, consiste à chauffer un mélange
d'aldéhyde et
d'amine primaire en solution dans le benzène ou le toluène, en présence d'une
quantité
catalytique d'acide para-toluènesulfonique (APTS), en utilisant un montage à
reflux équipé d'un
séparateur de Dean-Stark. Expliquer l'intérêt de cette technique et le
phénomène mis à profit.
12
3. Déterminer la configuration absolue de la lysine naturelle [1]. Indiquer et
justifier la structure de
l'imine [6] issue de la condensation du groupe amino de la lysine en position 2
et du phosphate de
pyridoxal [5] au pH physiologique (milieu considéré comme neutre).
4. Au sein du site actif de l'enzyme, l'imine [6] formée subit une
décarboxylation conduisant à un
anion intermédiaire [7]. À partir de l'étude de la structure de [7], justifier
la grande facilité avec
laquelle cette décarboxylation se produit.
À partir de l'intermédiaire [7], deux voies de biosynthèse sont envisageables
pour accéder au 5aminopentanal [3] qui se cyclise ensuite spontanément en
#1-pipéridéine [4]. Il est possible d'envisager
selon une première voie la transformation de [7] en [3] sans passage par la
cadavérine [2].
5. Montrer que la protonation de [7] peut conduire à un composé intermédiaire
[8] dont l'hydrolyse
engendre directement le 5-aminopentanal [3] ainsi qu'un produit [9] à partir
duquel le cofacteur
doit être régénéré. Donner les structures des produits [8] et [9].
Une deuxième voie de biosynthèse, en accord avec le Schéma 1, envisage le
passage par cadavérine
[2] puis son oxydation en [3].
6. Proposer un mécanisme expliquant la formation en une étape de [2] à partir
de [7] ainsi que le
rôle catalytique du phosphate de pyridoxal [5].
7. Expliquer pourquoi la transformation de [2] en [3] est une oxydation, et
pourquoi elle ne
s'effectue que sur l'un des groupements amino.
L'oxydation d'amines de formule générale R''CH2NH2 en aldéhydes R''CHO
peut être aussi catalysée par des enzymes (amine-oxydases à cuivre) qui
utilisent
comme cofacteur la topaquinone pour laquelle on adoptera la formule simplifiée
[10] ci-contre (R1 = reste polypeptidique).
8. Écrire la structure de l'imine [11] qui résulterait de la condensation (au pH
physiologique) de la cadavérine [2] et de la topaquinone [10], en justifiant
lequel des groupes carbonyle a ou b est impliqué.
OH
a
O
O
b
[10]
CH 2R1
9. L'imine [11] est en équilibre tautomérique avec une autre imine [12],
possédant un cycle
aromatique, dont la formation peut être catalysée par une base. Représenter
cette imine [12].
10. Quelle réaction l'imine [12] doit-elle subir pour conduire à [3] ? Indiquer
la structure du sousproduit [13] formé dans cette réaction, permettant la
régénération du cofacteur [10] par oxydation
([13] + O2 + H2O ( [10] + NH3 + H2O2).
11. En utilisant comme nutriment de la lysine dont les atomes de carbone en
position 2 ou 6 ont été
sélectivement marqués au carbone 14, il a été observé que les alcaloïdes
produits par les végétaux
correspondants incorporaient un cycle azoté à six chaînons sélectivement marqué
au carbone 14
en position 2 ou 6, respectivement. En déduire une information sur la véritable
voie de
biosynthèse de [4] à partir de la lysine [1].
II.
Synthèse de l'allosédamine
La ()-allosédamine [14] appartient à la famille des alcaloïdes de lobelia
inflata
Me
extraits d'une plante (« tabac indien ») poussant sur le continent
nord-américain et
OH N
utilisée par les populations locales, au dix-neuvième siècle, pour traiter les
problèmes respiratoires malgré sa toxicité à dose élevée. Le cycle azoté à six
Ph
[14]
chaînons de l'allosédamine est issu du métabolisme de la lysine alors que la
chaîne
(
)-Allosédamine
latérale provient d'un autre aminoacide, la phénylalanine. Une synthèse de la
()allosédamine [14] a été réalisée à partir de l'alcool énantiomériquement
enrichi
[15] (F.-X. Felpin et J. Lebreton, 2002) ; le début de cette synthèse est
décrit dans le Schéma 3.
13
OH
m-CPBA
Ph
[18]
CH2Cl2, 0 °C
[15]
+
[18']
([18]/[18'] = 1/1)
Cl
K2CO3, MeOH, 20 °C
O
O
O
O
O
IBr
Ph
O
m-CPBA
I
Ph
toluène, -85 °C
[16]
CO3H
+
+
HBr
[17]
Schéma 3 Début de la synthèse de la ()-allosédamine
Le carbonate mixte [16] (synthétisé à partir de l'alcool [15]) réagit d'abord
avec le bromure d'iode dans
le toluène à -85 °C pour conduire sélectivement au carbonate cyclique [17].
12. Proposer un mécanisme pour la formation de [17] à partir de [16] (il n'est
pas demandé de
justifier la stéréosélectivité).
Dans le spectre de RMN du proton du composé [17] (enregistré à 200 MHz dans
CDCl3), les
signaux suivants sont observés :
Déplacement chimique
7,48 7,28 ppm
Multiplicité du signal observé, constantes de couplage
Intégration
multiplet
5H
5,49 ppm
doublet de doublets, J = 11,9 Hz et J = 2,9 Hz
1H
4,65 ppm
multiplet
1H
3,47 ppm
doublet de doublets, J = 10,7 Hz et J = 4,4 Hz
1H
3,33 ppm
doublet de doublets, J = 10,7 Hz et J = 7,3 Hz
1H
2,65 ppm
doublet de triplets, J = 14,3 Hz et J = 2,9 Hz
1H
2,04 ppm
doublet de triplets, J = 14,3 Hz et J = 11,7 Hz
1H
Ces caractéristiques spectrales permettent d'attribuer les orientations
relatives des substituants présents
sur le cycle à six chaînons B du composé [17] représenté en Figure 1 dans sa
conformation chaise la
plus stable en respectant la numérotation des atomes. Pour l'analyse du
spectre, vous disposez des
tables de déplacement chimiques en annexe et des valeurs représentatives
données dans la Figure 1.
O
1HO
Ph
O H3
C2
C1
2'
4/4'
H a2
O
O
I
C3
H HH H
2
C1
C2
H e1
O
C3
H e2
B
H a1
[17]
2J(H
a1-He1)
= 1215 Hz
3J(H
a1-Ha2)
= 1012 Hz
3J(H
a1-He2)
= 25,5 Hz
3J(H
e1-He2)
= 24 Hz
Figure 1 Représentation du cycle B de [17] et valeurs représentatives des
constantes de couplage
entre des protons en position équatoriale/axiale portés par le même carbone
(2J) ou par deux carbones
voisins (3J) sur un cyclohexane en conformation chaise.
13. À l'aide de la table des J données en Figure 1 et de la table de
déplacements chimiques fournie en
annexe, attribuer le déplacement chimique du signal correspondant à H-1 et en
déduire la position
équatoriale ou axiale du groupe phényle sur le cycle B de [17].
14. Positionner sur une représentation dans l'espace de [17], dessiné dans sa
conformation B, les
protons H-1, H-2, H-2' et H-3. Attribuer les déplacements chimiques des signaux
correspondant à
14
H-2, H-2' et H-3. Déduire de l'examen des constantes de couplage de ces protons
la position
équatoriale ou axiale du groupe iodométhyle.
15. Le traitement de [17] par le carbonate de potassium dans le méthanol mène
ensuite sélectivement
au composé [18]. Par ailleurs, l'alcool [15] a été traité par l'acide
méta-chloroperoxybenzoïque
(m-CPBA) dans le dichlorométhane à 0 °C et, dans ces conditions, un mélange
équimolaire des
deux composés isomères [18] et [18'] est obtenu. Indiquer la structure des
composés [18] et [18']
et préciser leur lien de stéréoisomérie.
La fin de la synthèse de la ()-allosédamine est résumée sur le Schéma 4.
[18]
t-BuMe2SiCl
Et 3N
TBSO
[19]
CH 2Cl 2, 20 °C
MeSO2Cl
Et 3N
OH
?
Ph
[21]
CH 2Cl 2, 0 °C
[20]
MeNH 2, H 2O
[22]
DMF, 50 °C
Boc2O, Et 3N
CH 2Cl 2, 20 °C
OSO 2Me
( )-Allosédamine
[14]
HCl, MeOH, 60 °C
Me
Boc
TBSO
N
Ph
Me
Boc
TBSO
N
?
Ph
[24]
O
TBS = t-BuMe2Si
Boc2O
O
[23]
O
O
O
Schéma 4 Fin de la synthèse de la ()-allosédamine
Le composé [18] est traité par le chlorure de tert-butyldiméthylsilyle en
présence de triéthylamine
puis le composé [19] obtenu est transformé en alcool secondaire [20]. L'alcool
[20] traité par le
chlorure de méthanesulfonyle en présence de triéthylamine conduit au composé
[21] (C20H34O4SSi) qui
est ensuite chauffé avec une solution aqueuse de méthylamine dans le
diméthylformamide (DMF) pour
obtenir l'amine [22]. Après traitement de [22] par le dicarbonate de
di-tert-butyle (Boc2O), le carbamate
de tert-butyle [23] est isolé puis transformé en composé [24]. Finalement, le
chauffage de [24] en
présence d'acide chlorhydrique concentré dans le méthanol à 60 °C s'accompagne
d'un dégagement
gazeux de dioxyde de carbone et de 2-méthylpropène (l'éther silylé est aussi
coupé dans ces conditions
fortement acides). Après ajout d'hydrogénocarbonate de sodium solide jusqu'à
obtention d'un pH de
9-10 puis extraction par le dichlorométhane et purification par chromatographie
sur colonne de silice, la
()-allosédamine [14] est isolée.
16. Préciser l'intérêt de la réaction menant à [19] dans la synthèse.
17. Proposer un réactif pour transformer [19] en alcool secondaire [20].
18. Indiquer les structures de [21] et de [22] en précisant bien la
configuration de leurs centres
stéréogènes.
19. Suggérer une séquence de plusieurs étapes permettant de transformer [23] en
[24].
20. Expliquer la transformation de [24] en [14]. Expliquer pourquoi il est
nécessaire lors du
traitement final d'amener le mélange réactionnel à pH 10 avant extraction.
Une synthèse de la (+)-allosédamine utilise comme intermédiaire-clé le composé
[ent-18],
énantiomère de [18], comme indiqué dans le Schéma 5 en page suivante (B. Kang
et S. Chang, 2004).
15
ent-18
MeOCH2Cl
i-Pr2NEt
OMe
Me
N
O
?
25
27 (0.1 équiv)
Ph
CH2Cl2, 20 °C
28
C6H6, reflux
26
40%
PCy3 H
Cl
Cy = cyclohexyle
Ru
Cl
Ph
PCy3
27
1) H2, PtO2 cat.
CH3CO2Et, 20 °C
2) HCl aqueux
MeCN, 20 °C
3) NaHCO3 (pH 9-10)
(+)-Allosédamine
(14)
Schéma 5 Synthèse de la (+)-allosédamine à partir de [ent-18]
Ce dernier est traité par le chlorure de méthoxyméthyle en présence d'une amine
tertiaire puis le
composé [25] obtenu est transformé en amine tertiaire [26]. Une solution du
composé [26] est ensuite
traitée par le complexe de ruthénium [27] (0,1 équiv) et chauffée dans le
benzène à reflux pour conduire
à nouveau un composé [28] (isolé avec un rendement moyen de 40%). Le composé
[28] est alors
successivement traité par du dihydrogène en présence d'une quantité catalytique
d'oxyde de platine puis
par de l'acide chlorhydrique pour obtenir, après passage en milieu basique,
l'allosédamine [14].
21. En s'inspirant de la synthèse précédente et en conservant la méthylamine
comme source de
nucléophile azoté, proposer une suite de réactions permettant de transformer
[25] en amine
tertiaire [26].
22. En s'inspirant du cycle catalytique donné ci-contre, où
[Ru]=CH2 représente le complexe de ruthénium, indiquer la
structure de [28] obtenu à partir de [26]. Identifier le sousproduit gazeux
formé. Préciser si le complexe de ruthénium
[27] est un catalyseur ou un précurseur de catalyseur pour
cette réaction.
23. Expliquer pourquoi la déprotection en milieu acide aqueux
de l'alcool du composé issu de l'hydrogénation catalytique
de [28] est aisée.
III.
Synthèse totale de la fawcettimine
La fawcettimine [29], isolée pour la première fois en 1959, appartient à la
famille des alcaloïdes de
lycopodium qui possèdent des structures polycycliques originales, et sont des
inhibiteurs de
l'acétylcholinestérase. Ces alcaloïdes sont en partie issus du métabolisme de
la lysine. Ainsi, la
fawcettimine [29] possède une structure tétracyclique et un motif carbinolamine
qui est en équilibre
avec une forme ouverte dicéto-amine tricyclique [30] (Schéma 6).
O
HO
Me
H
Me
9
5
7
Me
=
N
Fawcettimine
11
HO
13
12
4
O
H
1
H
9
A 12 7 B
4
11
O
N15
[29]
15
[30]
13
5
O
H
C
N
H
1
Schéma 6 Équilibre de la fawcettimine avec sa forme ouverte
Dans cette partie, une synthèse totale de la fawcettimine est étudiée (G. Pan
et R. M. Williams, 2012).
Elle débute par l'élaboration du cycle B de [30] à partir du bromocétal [31]
selon le Schéma 7 (page
suivante).
16
O
O
t-BuOK
Br
DMSO
[32]
HO
Isoprène
CSA (0,02 %)
LiClO 4, Et 2O
O
?
Me
DMSO=diméthylsulfoxyde
O
KOH
H
[36]
H 2O, reflux, 15 h
H
[33] + [34]
[31]
O
O
Me
O [35]
C12 H16 O3
([33]/[34] = 95/5)
CSA
O
SO3H
Schéma 7 Construction du cycle B de la fawcettimine
24. Indiquer quel type réaction permet de passer de [31] à [32]. Donner la
structure de [32]. Le
composé [32] obtenu réagit avec l'isoprène (2-méthylbuta-1,3-diène) en présence
d'une quantité
catalytique d'un acide de Brønsted tel que l'acide camphre-10-sulfonique, noté
CSA, dans une
solution concentrée de perchlorate de lithium dans l'éther anhydre, et conduit
à un mélange de
deux composés [33] et [34] dans un rapport 95/5.
25. Quel type de réaction permet la synthèse du composé [33] ? Quelle espèce,
engendrée à partir de
[32] et possédant une double liaison appauvrie en électrons, participe à cette
réaction ? On précise
que le milieu LiClO4/Et2O favorise la réaction mais que celle-ci n'a pas lieu
en l'absence de CSA.
En outre, le même résultat est obtenu quel que soit l'énantiomère de CSA
utilisé.
26. Indiquer la structure du produit minoritaire [34].
27. Comment peut-on transformer [33] en [35] ?
28. Le spectre de RMN 1H (300 MHz) du composé [36] en solution dans CDCl3
présente les signaux
suivants : ! (ppm) 6,61 (multiplet, 1H), 4,00-3,77 (multiplet, 4H), 3,50
(multiplet, 1H), 2,78-2,48
(multiplet, 3H), 2,28 (singulet, 3H), 2,00-1,51 (multiplet, 4H). Indiquer la
structure de [36] en la
justifiant et proposer un mécanisme pour sa formation à partir de [35].
Le bicycle A-B de [30] est ensuite élaboré à partir de [36] en accord avec le
Schéma 8.
29. La cycloaddition de [36] et [37], réalisée en tube scellé à 180 °C, est
totalement régiosélective.
Indiquer la structure du composé majoritaire [38] obtenu, sachant qu'il résulte
d'une
cycloaddition avec « approche endo ». Quel est le lien de stéréoisomérie entre
[38] et [38'] ?
Me
(4 équiv)
[37]
OSiMe3
[36]
180 °C
[38]
+
[38']
A
1) n-Bu4NF
CH 2Cl 2
Me
H
2) Oxydation
A
B
O
H
i) H 2, Pd/C cat.
AcOEt
O
O
B
H
Me
ii) DBU (1 équiv)
iii) H 2, Rh/Al 2O3 cat.
B
O
RO
H
MeSO 2Cl
pyridine
H
Me
1) (Me 3Si)2NLi
O
THF, -78 °C
2) O
A
H
Me
m-CPBA
O
CO 2Me
[42] R = H
CH 2Cl 2
O
O
O
[40] CO Me
2
OMe
O
HO
B
H
CN
H
A
CO 2Me
[44]
HO
O H
Me
O
[39]
([38]/[38'] = 70/30)
H
H
Me
1) NaBH 4
MeOH
CH 2Cl 2
-40 °C
H
A
B
O
HO
H
2) HCl/H 2O
acétone
O
CO 2Me
[41]
[43]
1
N
DBU
8
N
Schéma 8 Construction du bicycle A-B de la fawcettimine
30. L'énone [39] qui résulte d'une déprotection suivie d'une oxydation du
mélange précédent [38] +
[38'] génère par traitement avec (Me3Si)2NLi un intermédiaire qui est opposé au
cyanoformiate
17
de méthyle pour donner [40]. Quelle espèce est engendrée à partir de [39] ? Le
composé [40]
possède deux groupes carbonyle cétoniques. Expliquer lequel est le plus réactif
vis-à-vis d'une
attaque nucléophile.
31. La transformation de [41] (obtenu sous forme d'un mélange de
diastéréoisomères par réduction
de [40] suivie d'une hydrolyse) en [42] est induite par traitement avec l'acide
méta-chloroperoxybenzoïque (m-CPBA). Pourquoi la double liaison C=C de [41]
n'est-elle pas affectée lors de cette
étape ?
Le composé [43] obtenu en traitant [42] par le chlorure de méthanesulfonyle
dans la pyridine, est
engagé dans une séquence de trois étapes successives (réalisées dans le même
réacteur) : hydrogénation
catalysée par du palladium sur charbon dans l'acétate d'éthyle, addition de
DBU, puis hydrogénation
catalysée du produit résultant par du rhodium sur alumine.
32. Quel est le rôle de chacune des étapes menant de [43] à [44] ? Indiquer
quel atome d'azote du
DBU (N1 ou N8) possède des propriétés basiques (pKa du couple DBU·H+ / DBU =
12,4), et
justifier le raisonnement utilisé.
La construction finale du tricycle A-B-C de [30] est assurée par la série de
transformations du
Schéma 9.
H
Me
Me
A
B
O
H
LiAlH4
(3 équiv)
O
O
[44] CO Me
2
H
9
5
7
12
O
4
11
O
13
1) (CH3CO)2O
(8 équiv), pyridine
[45]
THF, reflux
PhSH
KOH
MeCN
reflux
15
A
2) Catalyseur d'Otera
MeOH/THF
Me
H
Me
9
5
7
12
O
4
11
O
[48]
N
Ns
Ac = COCH3
AcO
OH
[46]
OH
H
9
A12 7 B
?
AcO
13
OAc
B
5
NsNH2 (4 équiv)
Ph3P (6 équiv)
DEAD (6 équiv)
MeCN/pyridine
OAc
4
11
15
1
N
H
1) HBr anhydre, CH2Cl2
2) K2CO3
[49]
H
Me
H
15
1
[47]
13
C
N
Ns
CO2Et
N N
1
EtO2C
DEAD
SO2NH2
Fawcettimine
NO2
NsNH2
Schéma 9 Élaboration du tricycle A-B-C et obtention de la fawcettimine
33. Le composé [44] subit une réduction exhaustive par LiAlH4. Indiquer la
structure de [45].
Combien d'équivalents de LiAlH4 seraient théoriquement nécessaires pour cela ?
Le polyol obtenu [45] est ensuite estérifié par action d'un large excès
d'anhydride éthanoïque dans
la pyridine pour conduire à un tétra-ester dont les deux groupes hydroxy
primaires sont sélectivement
libérés par traitement avec le catalyseur d'Otera [t-Bu2Sn(OH)Cl]2 dans un
mélange MeOH/THF. Le
cycle azoté à neuf chaînons (cycle C) est construit en traitant le diol [46]
par le 2-nitrobenzènesulfonamide (NsNH2) en présence de triphénylphosphine et
d'azodicarboxylate de diéthyle (DEAD).
34. Sachant qu'en présence de triphénylphosphine, d'azodicarboxylate de
diéthyle (DEAD) et d'une
substance acide A-H [pKa(AH/A-) < 13], un alcool R1CH2OH conduit à un sel d'oxyphosphonium de formule R1CH2-O-PPh3+·A- (l'hydrazide EtO2C-NH-NH-CO2Et étant formé comme sousproduit), proposer un mécanisme pour expliquer la formation du cycle azoté du composé [47]. 35. Quelles sont les opérations à réaliser pour transformer le tricycle [47] en dicétone [48] ? 18 36. Le groupement 2-nitrobenzènesulfonyle (Ns) présente l'intérêt de pouvoir être coupé par traitement avec un excellent nucléophile tel le thiophénolate de potassium (Schéma 10). Cette réaction fait intervenir un mécanisme en deux étapes correspondant à une addition suivie d'une élimination. Proposer une structure pour l'intermédiaire C et en déduire le rôle important exercé par le groupe nitro. Ecrire la structure de l'intermédiaire D. O R2 S N 3 O R NO2 PhSK C D SPh R2 H2O N H R3 SO2 NO2 Schéma 10 Étapes de coupure d'une 2-nitrophénylsulfone par le thiophénolate de potassium Après déprotection de l'atome d'azote, l'aminodicétone [49] n'est pas isolée mais directement traitée par une solution de HBr dans le dichlorométhane anhydre pour conduire à la formation de cristaux de bromhydrate d'ammonium de la fawcettimine, à partir desquels l'alcaloïde est libéré par traitement avec K2CO3. 37. La configuration du carbone C4 est différente dans le composé [49] et dans la fawcettimine. Il a été observé que cette inversion de configuration se produisait au moment du traitement de [49] en milieu acide (HBr) qui conduit à la formation du squelette tétracyclique de [29]. Par quelle espèce intermédiaire cette inversion de configuration se produit-elle ? La représenter. 38. La fawcettimine obtenue à l'issue de cette synthèse est-elle obtenue sous forme optiquement enrichie ou sous forme de mélange racémique ? Données : Tables de spectroscopie RMN 1H Déplacements chimiques Déplacement chimique Substituant (en ppm) C=C-H 4,5 7,0 CH2-C=C 2,1 2,6 CH-C=C 2,4 2,9 Ar-H 6,5 8,2 CH-Ar 2,4 2,6 CH2-I 2,1 3,5 CH2-C-O (cyclique) 1,5 1,7 CH-O (cyclique) 3,8 5,3 C6H5-CH-O 4,4 4,9 CH3-CO2,1 2,3 ! ! ! 19 pKa de la lysine pKa1 = 2,2 pKa2 = 8,9 pKa3 = 10,5