
X/ENS Chimie PC 2022
| Thème de l'épreuve | Étude de la (-)-quinine et de la (+)-quinidine |
| Principaux outils utilisés | chimie organique, thermodynamique, solutions aqueuses, cinétique chimique, oxydoréduction, cristallographie |
| Mots clefs | fluorescence, riboflavine |
Corrigé
:👈 gratuite pour tous les corrigés si tu crées un compte
👈 l'accès aux indications de tous les corrigés ne coûte que 1 € ⬅ clique ici
👈 gratuite pour tous les corrigés si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - -
👈 gratuite pour ce corrigé si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Énoncé complet
(télécharger le PDF)


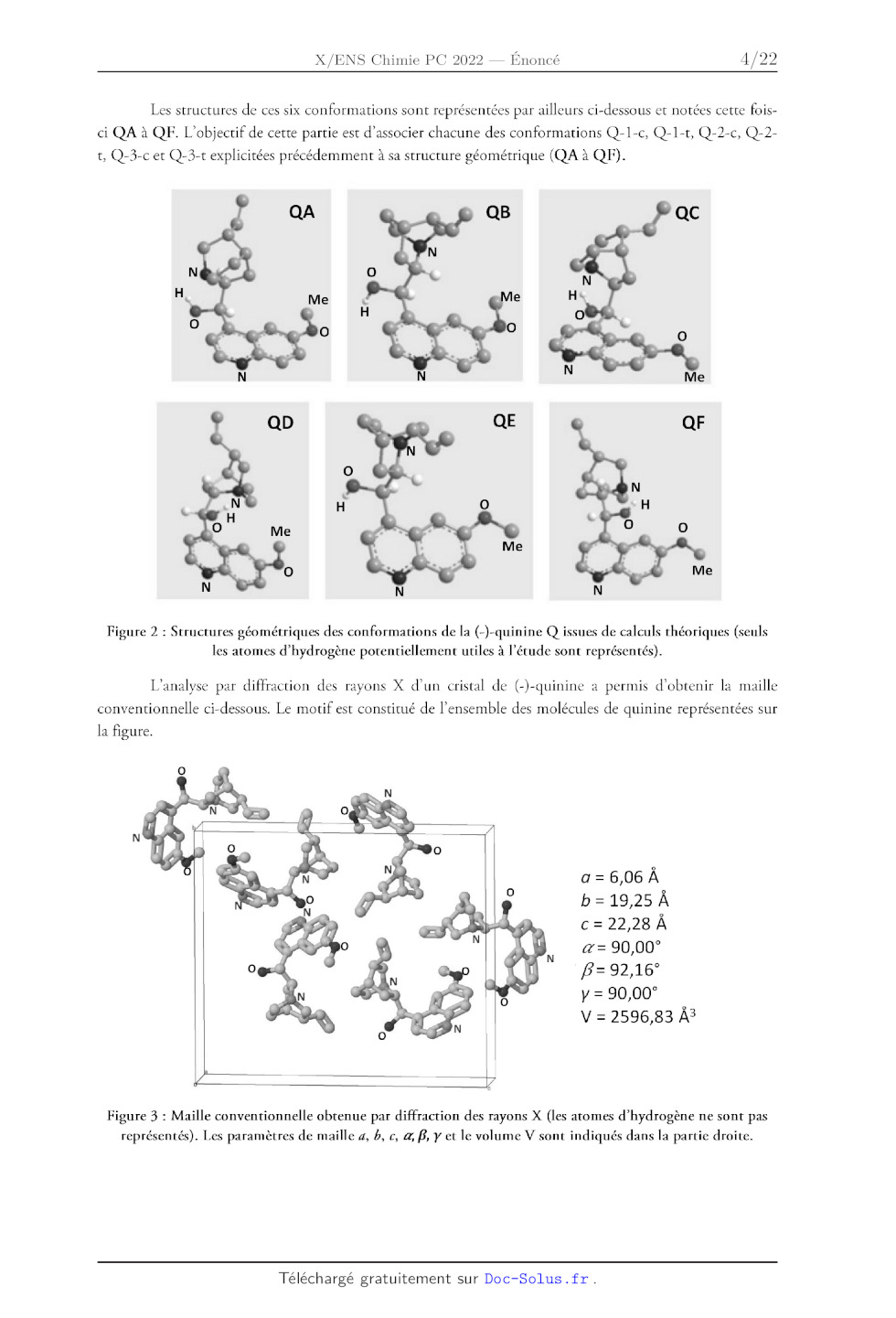










Rapport du jury
(télécharger le PDF)



Énoncé obtenu par reconnaissance optique des caractères
ECOLE POLYTECHNIQUE - ESPCI
ECOLES NORMALES SUPERIEURES
CONCOURS D'ADMISSION 2022
MARDI 26 AVRIL 2022
14h00 - 18h00
FILIERE PC - Epreuve n° 4
CHIMIE A (XEULS)
Durée : 4 heures
L'utilisation des calculatrices n'est pas autorisée pour cette
épreuve
Étude de la (-)-quinine et de la (+)-quinidine
La (-)-quinine et la (+)-quinidine appartiennent à la famille des alcaloïdes et
possèdent des propriétés
thérapeutiques importantes. Leurs structures sont représentées sur la figure
ci-dessous. Elles ont des
propriétés basiques et sont constituées d'hétérocycles azotés : l'un de type
quinoline portant un groupement
méthoxy et l'autre de type quinuclidine portant un groupe fonctionnel vinyle
(-CH=CH:). Ces molécules
sont stéréoisomères l'une de l'autre et leur relation est qualifiée de
pseudo-énantiomérie. Ces deux espèces
chimiques ont été originellement extraites de l'écorce de l'arbuste quinquina
dans les années 1820-30 par
des chimistes ou pharmaciens français. Historiquement, cette écorce était
notamment utilisée dès le début
du XVII siècle dans la cordillère des Andes comme antipyrétique (lutter contre
la fièvre). Des études
ultérieures ont montré que ces propriétés sont propres à la (-)-quinine et que
celle-ci possède en complément
des propriétés analgésiques et antipaludiques. La (+)-quinidine pour sa part a
montré des propriétés
antiarythmiques intéressantes. La quinine est également un composant
aromatisant car elle possède un goût
amer. Elle est par exemple présente dans les boissons qualifiées de « Tonic ».
LUN _ ï
-- S
LL < ' Ho S US : Hétérocycle quinuclidine ; H,CO ÈS H,CO Ki N : N ï (-)-quinine (+)-quinidine ! Hétérocycle quinoline : Figure 1 : Structures de la (-)-quinine et de la (+)-quinidine. Ce sujet propose de s'intéresser dans une première partie (partie A) à l'analyse structurale (configuration et conformation) de la (-)-quinine et de la (+)-quinidine. La deuxième partie du sujet (partie B) est dédiée à l'étude et à la comparaison de quelques propriétés physico-chimiques de ces deux espèces chimiques (solubilité, propriétés acido-basiques et propriétés photophysiques) ainsi qu'à l'utilisation des propriétés de fluorescence de la (-)-quinine pour l'analyse quantitative. Finalement, la dernière partie (partie C) s'intéresse à la synthèse de ces deux espèces chimiques. Les différentes parties sont partiellement reliées mais présentent chacune un grand nombre de questions indépendantes. Les questions précédées d'un symbole (*) seront valorisées dans la notation car elles nécessitent plusieurs étapes de raisonnement et peuvent s'avérer plus longues à traiter. Des données physico-chimiques sur les molécules de (-)-quinine et (+)-quinidine sont reportées dans l'annexe 1. Des données spectroscopiques (IR et RMN) et thermodynamiques sont par ailleurs reportées : ÿ ; : . : . , 8 respectivement dans les annexes 2 et 3. Une banque de réactions de chimie organique fait finalement l'objet de l'annexe 4. Partie A. Étude structurale de la (-)-quinine et de la (+)-quinidine L. IL. 1. Stéréoisomérie de configuration Donner la signification du signe (-) dans le nom de la (-)-quinine. Dans le nom donné à la (-)-quinine en nomenclature IUPAC (annexe 1), indiquer le nom de la molécule considérée comme chaîne principale et des groupes fonctionnels considérés comme des substituants. Localiser sur la structure de la (-)-quinine, les carbones stéréogènes notés À, 25, 4R, 8R dans le nom en nomenclature IUPAC de la (-)-quinine. Justifier le descripteur stéréochimique (À) spécifié au début du nom en nomenclature [UPAC de la (-)-quinine. Donner le nombre maximum de stéréoisomères théoriquement possibles pour la (-)-quinine. Indiquer combien cette molécule en possède effectivement en justifiant pourquoi. (*) Indiquer la relation stéréochimique liant la (-)-quinine et la (+)-quinidine. Expliquer pourquoi on parle de relation de pseudo-énantiomérie entre ces molécules en vous appuyant sur les propriétés physico-chimiques données dans l'annexe 1. Stéréoisomérie de conformation Des calculs théoriques ont été menés récemment pour identifier les différentes conformations possibles pour la molécule de (-)-quinine. Six conformations particulières ont été identifiées et notées Q-1- c, Q-1-t, Q-2-c, Q-2-t, Q-3-c, Q-3-t. Leur énergie relative dans le vide ainsi que quelques angles dièdres et une valeur théorique du nombre d'onde de la vibration d'élongation OH sont reportés dans le tableau ci- dessous pour chaque conformation. L'orientation du groupement méthoxy (OMe) de l'hétérocycle quinoline relativement à l'hétérocyle quinuclidine est spécifiée dans le nom de la conformation par un c pour cis, si le méthyle de ce groupe est orienté vers l'hétérocyle quinuclidine, et t pour #rans, s'il ne l'est pas. Les conformations Q-1-c et Q-1-t ne diffèrent donc dans leur structure que par l'orientation du groupement méthoxy. Il en va de même pour Q-2-c et Q-2-t ainsi que Q-3-c et Q-3-t. Q-1-c Q-1-t Q-2-c Q-2- Q-3-c Q-3-t 0 99,3 153,6 -82,4 174,0 3766 5,23 99,6 155,8 -80,4 172,2 3767 7,25 99,8 -87,5 39,0 -24,1 3514 9,48 100,5 -88,8 37,7 -23,0 3507 15,58 -81,6 -90 41,3 -23,0 3513 19,24 -83,4 -87,5 43,8 -25,7 3545 Tableau 1 : Énergie relative dans le vide, angles dièdres et nombre d'onde de la vibration d'élongation de liaison O-H dans le cas des six conformations issues de calculs théoriques. La numérotation des atomes est spécifiée sur la Figure 4. Les structures de ces six conformations sont représentées par ailleurs ci-dessous et notées cette fois- ci QA à QF. L'objectif de cette partie est d'associer chacune des conformations Q-1-c, Q-1-t, Q-2-c, Q-2- t, Q-3-c et Q-3-t explicitées précédemment à sa structure géométrique (QA à QF). Figure 2 : Structures géométriques des conformations de la (-)-quinine Q issues de calculs théoriques (seuls les atomes d'hydrogène potentiellement utiles à l'étude sont représentés). L'analyse par diffraction des rayons X d'un cristal de (-)-quinine a permis d'obtenir la maille conventionnelle ci-dessous. Le motif est constitué de l'ensemble des molécules de quinine représentées sur la figure. nn. °w N je / EUR a = 6,06 À b = 19,25 À c = 22,28 À a= 90,00° B= 92,16° y = 90,00° V = 2596,83 À5 Figure 3 : Maille conventionnelle obtenue par diffraction des rayons X (les atomes d'hydrogène ne sont pas représentés). Les paramètres de maille 4, b, c, &, B, y et le volume V sont indiqués dans la partie droite. Loi de Hooke En spectroscopie d'absorption infrarouge, la loi de Hooke permet de relier le nombre d'onde o à des y. .. , . . 1 K grandeurs caractéristiques de la liaison selon l'équation suivante : o(AB) = == à mamB 2 1: : où U = est la masse réduite, #1 et m8 les masses atomiques des atomes À et B et £ la constante Mma+m ATMB de raideur de la liaison (AB) concernée. Dans cette formule, c est la célérité de la lumière dans le vide. 7. Justifier que les structures QA à QF sont des conformations de la même molécule. 8. Définir ce qu'est une liaison (ou un pont) hydrogène et indiquer comment évolue son énergie en fonction de paramètres géométriques pertinents entre les atomes mis en jeu. 9. Indiquer comment se traduit la présence d'un pont hydrogène sur le nombre d'onde de la vibration d'élongation de la liaison O-H. 10. (*) En vous appuyant sur la question 8 et le tableau 1, identifier parmi les structures QA à QF celles qui sont susceptibles de présenter une liaison hydrogène intramoléculaire (d'énergie intermédiaire) sachant que les atomes mis en jeu dans ce pont hydrogène (que l'on précisera en utilisant la numérotation de la figure ci-dessous) sont les mêmes pour toutes les structures. 22 -------- 23 13 17 14 20 21 19 18 Figure 4 : Numérotation adoptée pour les atomes de la (-)-quinine dans le cadre de la partie A.IL. Attention, celle-ci diffère de la numérotation IUPAC de la partie A.I 11. (*) Associer, à l'aide des deux questions précédentes et du tableau 1, les conformations Q-1-c à Q-3-t à leurs structures QA à QF. Justifier soigneusement votre réponse. 12. Sachant que les résultats issus des calculs théoriques et de la diffraction des rayons X (figure 3) sont en accord, indiquer parmi les 4 représentations de Newman suivantes (I à IV) dans l'axe de la liaison C12-C4 celle attribuable à la conformation la plus stable de la (-)-quinine en justifiant. 13. 14. 15. 16. 17. El C1 C10 Cio C10 Sachant que dans le cas de la (+)-quinidine la conformation la plus stable possède un angle dièdre (C10-C12-C14-N:5) d'environ 180°, proposer une représentation de Newman analogue à celle de la question précédente pour la (+)-quinidine. Identifier la nature des liaisons ou interactions responsables de la cohésion du cristal de (-)-quinine. En déduire le type de cristal. En vous aidant de l'annexe 1, montrer que le nombre Z de molécules de (-)-quinine par maille est cohérent avec les données cristallographiques de la figure 3. À l'aide des questions précédentes (parties I et Il), identifier et expliquer la (les) différence(s) majeure(s) observée(s) dans les spectres IR de la (-)-quinine et la (+)-quinidine donnés dans l'annexe 1. (*) De même, après avoir indiqué le signal attribuable au proton porté par le carbone C:2 dans les spectres RMN de la (-)-quinine et la (+)-quinidine donnés dans l'annexe 1, expliquer la différence de constante de couplage observée pour ce signal dans le cas des deux molécules en vous aidant de l'annexe 2 et des réponses aux questions précédentes. Partie B. Propriétés physico-chimiques de la (-)-quinine et application aux dosages. I. Solubilité 18. Expliquer en quoi la valeur de ZogP dont la définition est donnée en annexe 1 permet d'appréhender le caractère hydrophile ou hydrophobe (lipophile) d'une molécule selon le signe et la valeur absolue de LogP. 19. Interpréter la valeur de LogP dans le cas de la (-)-quinine. Comparer cette valeur à celle de la (+)-quinidine et justifier. IL. Propriétés acido-basiques La (-)-quinine est une dibase. La structure donnée en début d'énoncé correspond à la forme de la quinine la plus basique notée Q. Les autres formes sont notées QH* et QH>"*.
20. Représenter le diagramme de prédominance des différentes formes de la
(-)-quinine en
précisant la structure complète de chacune des formes et en vous appuyant sur
les annexes 1
et 3.
21. (*) En vous appuyant sur les résultats de l'étude conformationnelle
(questions 12 et 13) et
l'annexe 1, expliquer pourquoi les valeurs de pXA pour la (+)-quinidine et la
(-)-quinine sont
identiques et pourquoi celles de pK sont différentes.
22. Classer, en le justifiant, les valeurs de LogP pour Q, QH: et QH2** par
ordre décroissant.
IT. Propriétés photophysiques
Document 1 : Définitions sur la fluorescence
L'interaction de la lumière avec une molécule M dans son état fondamental (noté
Ss) peut conduire au
processus d'absorption de photons (de constante de vitesse #3). La molécule est
alors excitée et atteint un
état que l'on appelle S:. Elle est notée M*. À partir de cet état, plusieurs
processus de désexcitation peuvent
avoir lieu :
- Retour à l'état fondamental Sç par un processus non radiatif (flèche ondulée
sur la figure ci-dessous)
de constante de vitesse #1.
- Retour à l'état fondamental S par un processus radiatif (flèche droite sur la
figure ci-dessous) de
constante de vitesse £; : il s'agit d'un phénomène appelé fluorescence.
- Passage sur un autre état excité noté T selon un processus non radiatif de
constante de vitesse 3.
Chaque processus est assimilé à un acte élémentaire de constante de vitesse k:.
Les processus de
désexcitation de T; ne sont pas considérés ici.
Fluorescence
L
Type de processus
Non
) Radiatif IN radiatif
(émission de lumière et absorption )
On définit par ailleurs l'intensité de fluorescence /{5) comme la quantité de
matière de photons
(en mol) émise par unité de temps et par unité de volume de solution lors du
processus de désexcitation
radiative entre S; et So (de constante de vitesse &). Le temps caractéristique
de l'état S; est appelé temps de
déclin +. Finalement, le rendement quantique de fluorescence D, qui quantifie
l'efficacité de la fluorescence
à la suite de absorption d'un photon est défini comme :
nombre de photons émis par fluorescence ko
un nombre de photons absorbés | kitki+ks
Comme vu en partie B.IT, la (-)-quinine et la (+)-quinidine comportent
plusieurs formes acido-
basiques. Ces formes QH>*, QH' et Q possèdent des rendements quantiques de
fluorescence ®f respectifs
de 0,55, 0,50 et 0,01. On considérera que ces rendements ne dépendent pas de la
longueur d'onde
d'excitation.
a. Excitation stationnaire
Pour avoir accès expérimentalement au spectre d'émission et au rendement
quantique dans un
solvant donné et à une température donnée, on réalise une excitation
stationnaire grâce à une source
continue de lumière dont on fixe la longueur d'onde (appelée longueur d'onde
d'excitation 1). La quantité
de matière de photons absorbés par unité de temps et de volume est noté Zà. On
considère dans le cas de
l'excitation stationnaire que la quantité de molécules excitées est faible par
rapport à la quantité de molécules
en solution. Les spectres d'absorption et d'émission de fluorescence pour une
longueur d'onde d'excitation
A de 333 nm des différentes formes acido-basiques de la (-)-quinine en solution
aqueuse sont donnés ci-
dessous. Le spectre d'émission pour QH;"* est normalisé à 1 au maximum
d'émission.
1.6
Absorbance
1.4 Fluorescence
Absorbance
1.2 Fluorescence
Fluorescence
0.8
/Intensité de fluorescence ----
0.6
Absorbance
0.0
220 240 260 280 300 320 340 360 380 400 420 440 460 480 500 520 540 560 580 600
Longueur d'onde / nm
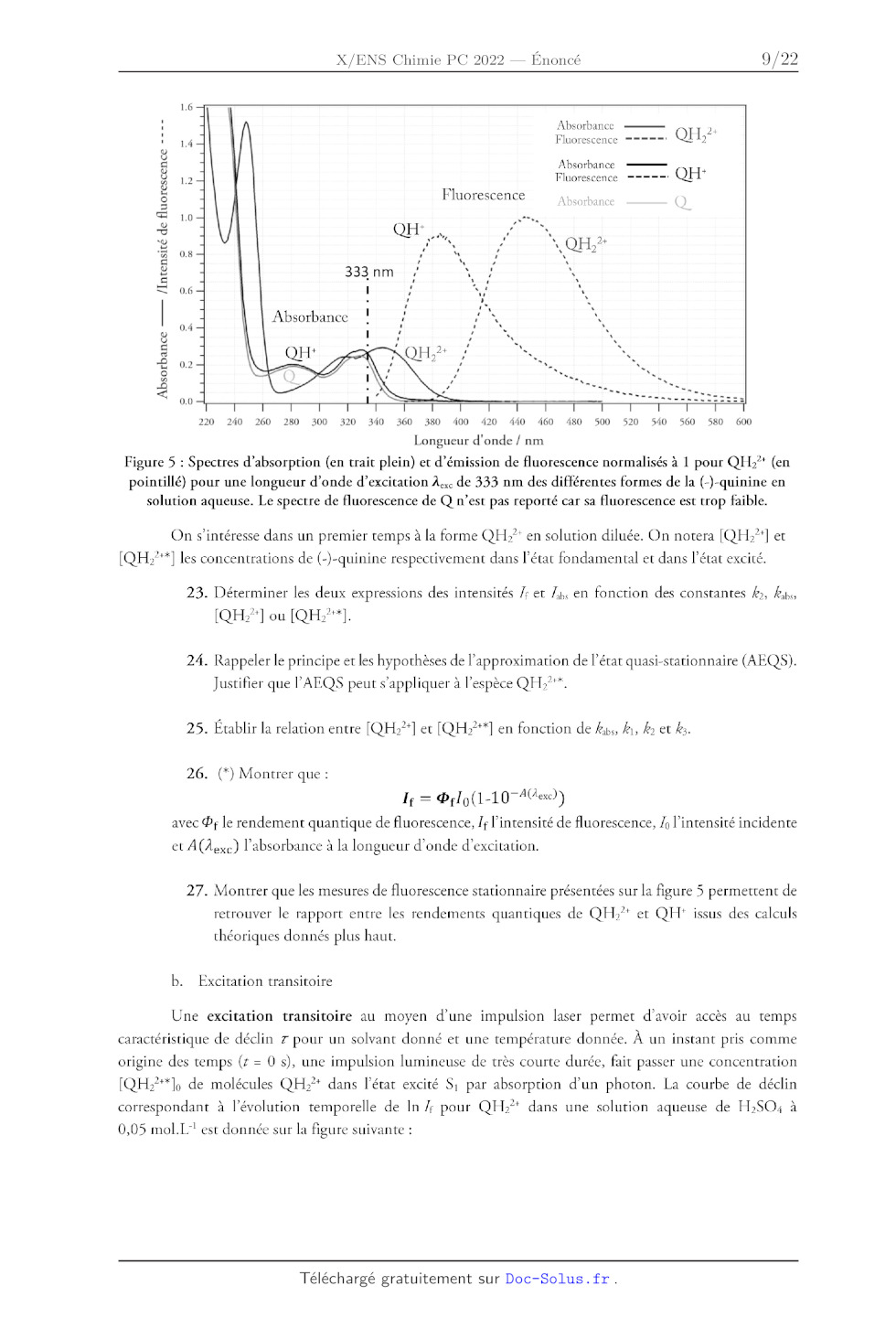
Figure 5 : Spectres d'absorption (en trait plein) et d'émission de fluorescence
normalisés à 1 pour QH;* (en
pointillé) pour une longueur d'onde d'excitation À. de 333 nm des différentes
formes de la (-)-quinine en
solution aqueuse. Le spectre de fluorescence de Q n'est pas reporté car sa
fluorescence est trop faible.
On s'intéresse dans un premier temps à la forme QH>* en solution diluée. On
notera [QH°] et
[QH27*] les concentrations de (-)-quinine respectivement dans l'état
fondamental et dans l'état excité.
23. Déterminer les deux expressions des intensités /; et Zà, en fonction des
constantes #, ka,
[QH:*] ou [QH:*].
24. Rappeler le principe et les hypothèses de l'approximation de l'état
quasi-stationnaire (AEQS).
Justifier que l'AEQS peut s'appliquer à l'espèce QH:**.
25. Établir la relation entre [QH2?*] et [QH2*] en fonction de ki, 41, 42 et k3.
26. (*) Montrer que :
I; = Pfl0(1-1074Cexc))
avec Df le rendement quantique de fluorescence, /f l'intensité de fluorescence,
1, l'intensité incidente
et A(Zexc) l'absorbance à la longueur d'onde d'excitation.
27. Montrer que les mesures de fluorescence stationnaire présentées sur la
figure 5 permettent de
retrouver le rapport entre les rendements quantiques de QH;"* et QH: issus des
calculs
théoriques donnés plus haut.
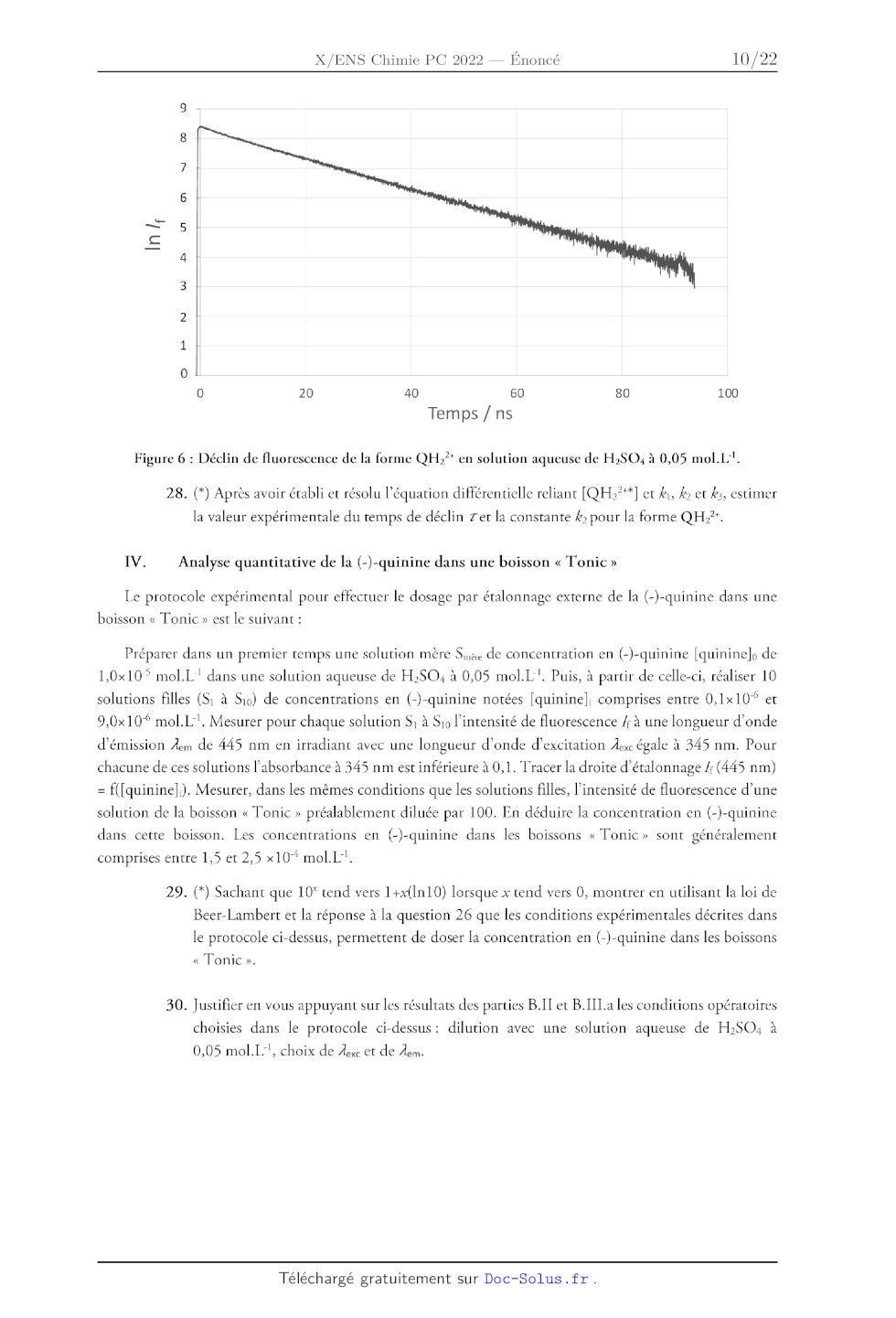
b. Excitation transitoire
Une excitation transitoire au moyen d'une impulsion laser permet d'avoir accès
au temps
caractéristique de déclin 7 pour un solvant donné et une température donnée. À
un instant pris comme
origine des temps (7 = 0 s), une impulsion lumineuse de très courte durée, fait
passer une concentration
[QH>7*] de molécules QH;7 dans l'état excité S1 par absorption d'un photon. La
courbe de déclin
correspondant à l'évolution temporelle de In Z: pour QH>"* dans une solution
aqueuse de H2SO4 à
0,05 mol.L'' est donnée sur la figure suivante :
In /
0 20 40 60 80 100
Temps /ns
Figure 6 : Déclin de fluorescence de la forme QH:"* en solution aqueuse de
H,SO: à 0,05 mol.L'.
28. (*) Après avoir établi et résolu l'équation différentielle reliant [QH2?*]
et k, k et k;, estimer
la valeur expérimentale du temps de déclin Zet la constante k; pour la forme
QH?.
IV. Analyse quantitative de la (-)-quinine dans une boisson « Tonic »
Le protocole expérimental pour effectuer le dosage par étalonnage externe de la
(-)-quinine dans une
boisson « Tonic » est le suivant :
Préparer dans un premier temps une solution mère Sa. de concentration en
(-)-quinine [quininelo de
1,0x10° mol.L' dans une solution aqueuse de H2SO: à 0,05 mol.L''. Puis, à
partir de celle-ci, réaliser 10
solutions filles (Si à S:v) de concentrations en (-)-quinine notées [quininel;
comprises entre 0,1x10% et
9,0x10% mol.L'. Mesurer pour chaque solution S; à Si l'intensité de
fluorescence /; à une longueur d'onde
d'émission em de 445 nm en irradiant avec une longueur d'onde d'excitation Aex
égale à 345 nm. Pour
chacune de ces solutions l'absorbance à 345 nm est inférieure à 0,1. Tracer la
droite d'étalonnage 7 (445 nm)
= f([quinine];). Mesurer, dans les mêmes conditions que les solutions filles,
l'intensité de fluorescence d'une
solution de la boisson « Tonic » préalablement diluée par 100. En déduire la
concentration en (-)-quinine
dans cette boisson. Les concentrations en (-)-quinine dans les boissons «
Tonic» sont généralement
comprises entre 1,5 et 2,5 x104 mol.L'.
29. (*) Sachant que 10* tend vers 1+x{(In10) lorsque x tend vers 0, montrer en
utilisant la loi de
Beer-Lambert et la réponse à la question 26 que les conditions expérimentales
décrites dans
le protocole ci-dessus, permettent de doser la concentration en (-)-quinine
dans les boissons
« Tonic ».
30. Justifier en vous appuyant sur les résultats des parties B.IT et B.IIL.a
les conditions opératoires
choisies dans le protocole ci-dessus: dilution avec une solution aqueuse de
H:SO: à
0,05 mol.L'!, choix de Ax et de Am.
10
31. Donner deux raisons permettant d'expliquer qu'il est préférable de diluer
par 100 la boisson
« Tonic». Proposer en conséquence, pour la boisson « Tonic», un solvant de
dilution
adéquat et identifier le paramètre clé à bien contrôler.
32. Indiquer s'il est possible de réaliser un dosage par étalonnage externe en
utilisant la valeur de
T plutôt que À. Justifier.
33. L'intensité de fluorescence à 445 nm pour une boisson « Tonic » diluée par
100 est de
318 u.a. (unité arbitraire). Sachant que la pente et l'ordonnée à l'origine de
la droite
d'étalonnage à 445 nm sont respectivement de 1,37x105 u.a.mol'.L et 18 u.a., en
déduire
une estimation de la concentration en (-)-quinine dans la boisson « Tonic » et
discuter de la
valeur obtenue.
V. Analyse quantitative de la riboflavine par inhibition de la fluorescence de
la (-)-quinine
La riboflavine (ou vitamine B2), dont la structure est représentée ci-après,
est essentielle à la
croissance et au développement des cellules. Sa concentration dans le sang doit
être contrôlée car la majeure
partie de cette vitamine est excrétée dans l'urine. Cette molécule ne peut pas
être synthétisée dans le corps
humain. Elle doit donc provenir de sources alimentaires telles que le foie, le
fromage, le lait, la viande, les
oeufs, le vin et le thé. Une carence en vitamine B2 peut entraîner des
problèmes de santé qu'il est possible
de traiter à l'aide de comprimés contenant une quantité contrôlée de
riboflavine.
OH
Figure 7 : Structure de la riboflavine (RF).
Pour quantifier la concentration en riboflavine, il est possible d'utiliser la
(-)-quinine sous sa forme
QH". En effet, cette dernière peut, en milieu H,SO4 à 0,05 moL.L', s'associer
avec 7 molécules de
riboflavine pour former une espèce que l'on notera QRF. Cette transformation
est modélisée par l'équation
de réaction suivante :
QH2* +n RF = QRF
Elle conduit à une diminution d'intensité de fluorescence /; de la (-)-quinine
(inhibition) mais ne
modifie pas le rendement quantique de celle-ci. L'analyse de cette inhibition
permet d'avoir accès à la
stoechiométrie de l'espèce QREF (nombre 7 de molécules de riboflavine REF
interagissant avec la (-)-quinine)
et aux données thermodynamiques relatives à sa formation (constante
thermodynamique de formation K°,
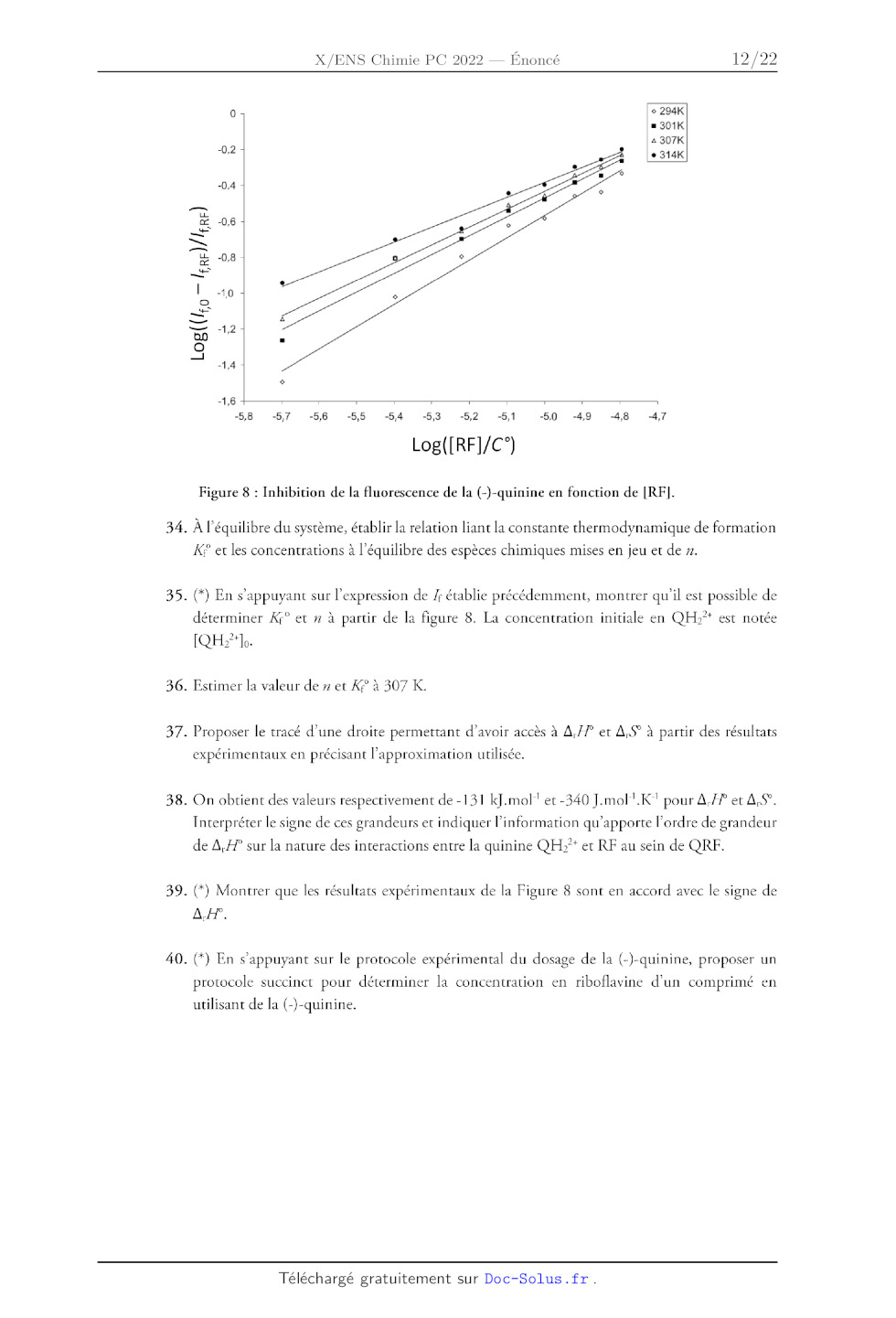
A,F, AS). Les résultats issus de cette analyse pour une excitation stationnaire
sont donnés sur la figure 8.
Dans cette partie, les intensités de fluorescence à 450 nm en l'absence et en
présence de riboflavine sont
respectivement notées 0 et /£rr. On considère également que le complexe n'émet
pas de lumière à la
longueur d'onde de 450 nm.
11
0 - o 294K
= 301K
à 307K
t? ° 314K
04 :
Can.)
Le
æ -06 |
--
S
----,
= 08.
ns
| 4.
Q,
--
TZ 12:
= À
O
=
1,4
©
1,6 : : : : : : : : : : |
5,8 57 -56 -55 54 -53 52 51 -50 -49 -48 47
Log([RF]/C°)
Figure 8 : Inhibition de la fluorescence de la (-)-quinine en fonction de [RE].
34. À l'équilibre du système, établir la relation liant la constante
thermodynamique de formation
KP et les concentrations à l'équilibre des espèces chimiques mises en jeu et de
».
35. (*) En s'appuyant sur l'expression de /; établie précédemment, montrer
qu'il est possible de
2+
déterminer Xf° et # à partir de la figure 8. La concentration initiale en QH:"*
est notée
[QH2* Jo.
36. Estimer la valeur de » et K;° à 307 K.
37. Proposer le tracé d'une droite permettant d'avoir accès à A, et A;S° à
partir des résultats
expérimentaux en précisant l'approximation utilisée.
38. On obtient des valeurs respectivement de -131 kJ.mol"' et -340 J.mol'.K'
pour A, et AS".
Interpréter le signe de ces grandeurs et indiquer l'information qu'apporte
l'ordre de grandeur
de A,7P sur la nature des interactions entre la quinine QH>* et RF au sein de
QRF.
39. (*) Montrer que les résultats expérimentaux de la Figure 8 sont en accord
avec le signe de
A.
40. (*) En s'appuyant sur le protocole expérimental du dosage de la
(-)-quinine, proposer un
protocole succinct pour déterminer la concentration en riboflavine d'un
comprimé en
utilisant de la (-)-quinine.
12
Partie C. Synthèses de la (-)-quinine et de la (+)-quinidine
Dans cette partie, un soin particulier est attendu dans l'écriture des
mécanismes réactionnels ; les
formules utilisées dans les réponses aux questions devront obligatoirement être
des représentations
topologiques et faire apparaître les doublets non liants et les formes
mésomères pertinentes des intermédiaires
réactionnels s'il y a lieu. Une représentation simplifiée des molécules peut
être utilisée dans l'écriture des
mécanismes réactionnels ou des équations de réaction mais, si c'est le cas,
elle doit être clairement explicitée.
Une banque de réactions de chimie organique est donnée en annexe 4.
Depuis la première synthèse de la quinine en 1918 à partir de la quinotoxine
obtenue par dégradation
de la quinine naturelle, plusieurs synthèses ont été proposées par la
communauté scientifique. Une des
dernières en date est celle qui sera étudiée dans cette partie. Elle utilise
comme précurseur la benzylamine A.
La première étape de la synthèse consiste à obtenir le composé D selon la
séquence réactionnelle 1
décrite ci-dessous. Le passage de la benzylamine A au composé B s'effectue en 2
étapes, un composé AB est
formé lors de la première étape. Ce dernier est alors engagé dans une deuxième
étape qui permet de remplacer
le groupement Bn par le groupement Cbz.
1. K2CO:, DMF O 0
BR ORZ OsO,(cat), HO° NalO4 Si, L
CT % _ Ga) ES T J K3[Fe(CN)ç] c CH>Cl
> >
2. CbzCl,toluène FBUOHGg (190%) \
Benzylamine 70°C °
A ë D
(80%) (100%)
Données
.
7°
Séquence réactionnelle 1 : Synthèse du composé D.
Le mécanisme conduisant à la formation du composé EUR met en jeu le cycle
catalytique suivant :
ê 2H,0, 2HO°
, R
ST = Le R
H,0 h
: R'
HO o HT +H,0
Il» _
O0, = 006 [Os0,(0H),77 9%
H,0
2 Fe(CN)s*-- 2 Fe(CN)é
13
41. Représenter la structure du composé AB et proposer un mécanisme réactionnel
rendant
compte de sa formation.
On s'intéresse maintenant plus en détail au cycle catalytique permettant de
former le composé C.
42. Indiquer la position de l'osmium (colonne et période) dans la
classification périodique
sachant que sa configuration électronique abrégée est : [Xe] 4f115466s2.
43. Représenter un schéma de Lewis de OsO: et [OsO(OH).}-. Les électrons des
orbitales fne
seront pas pris en compte. Indiquer, en justifiant, leurs géométries
respectives.
44. Indiquer le nombre d'oxydation de l'osmium dans OsO:; et [OsO:(OH)4J".
Expliquer
pourquoi le nombre d'oxydation de l'osmium dans OsO: est particulièrement
courant.
45. Écrire la demi-équation électronique associée au couple du fer mis en jeu
dans le cycle
catalytique. Indiquer le rôle et l'intérêt de cette réaction dans le cycle
catalytique.
yuq q y. q
46. (*) Donner l'expression du potentiel standard d'oxydoréduction du couple du
fer mis en jeu
dans la réaction d'oxydation du cycle catalytique en fonction de E°(Fe**/Fe*)
et des pA des
complexes mis en jeu. Déterminer la valeur de ce potentiel et discuter de
celle-ci au regard de
la question précédente et de l'annexe 3.
q P
47. En vous appuyant sur le cycle catalytique, représenter une structure pour
le composé C.
Représenter les différents stéréoisomères de C formés et indiquer la relation
qui existe entre
EUX.
48. Indiquer une réaction s'apparentant à celle mise jeu lors du passage du
composé B au
composé D.
La synthèse se poursuit par l'obtention du composé I selon la séquence
réactionnelle 2 représentée ci-
dessous. Le composé D est mis à réagir dans le dichlorométhane CH;CL en
présence de proline (catalyseur)
pour former deux composés (E-cis et E-trans) par une réaction de
cycloaldolisation énantiosélective. La
formation du composé H à partir du composé G s'opère grâce à une réaction
d'oléfination de Peterson dont
le mécanisme est représenté sur la figure 9. H est obtenu sous la forme de 2
stéréoisomères. Le composé I
est formé en 2 étapes à partir du composé H. Le composé formé à l'issue de la
première étape est noté HI.
BzCI correspond au chlorure de benzoyle.
14
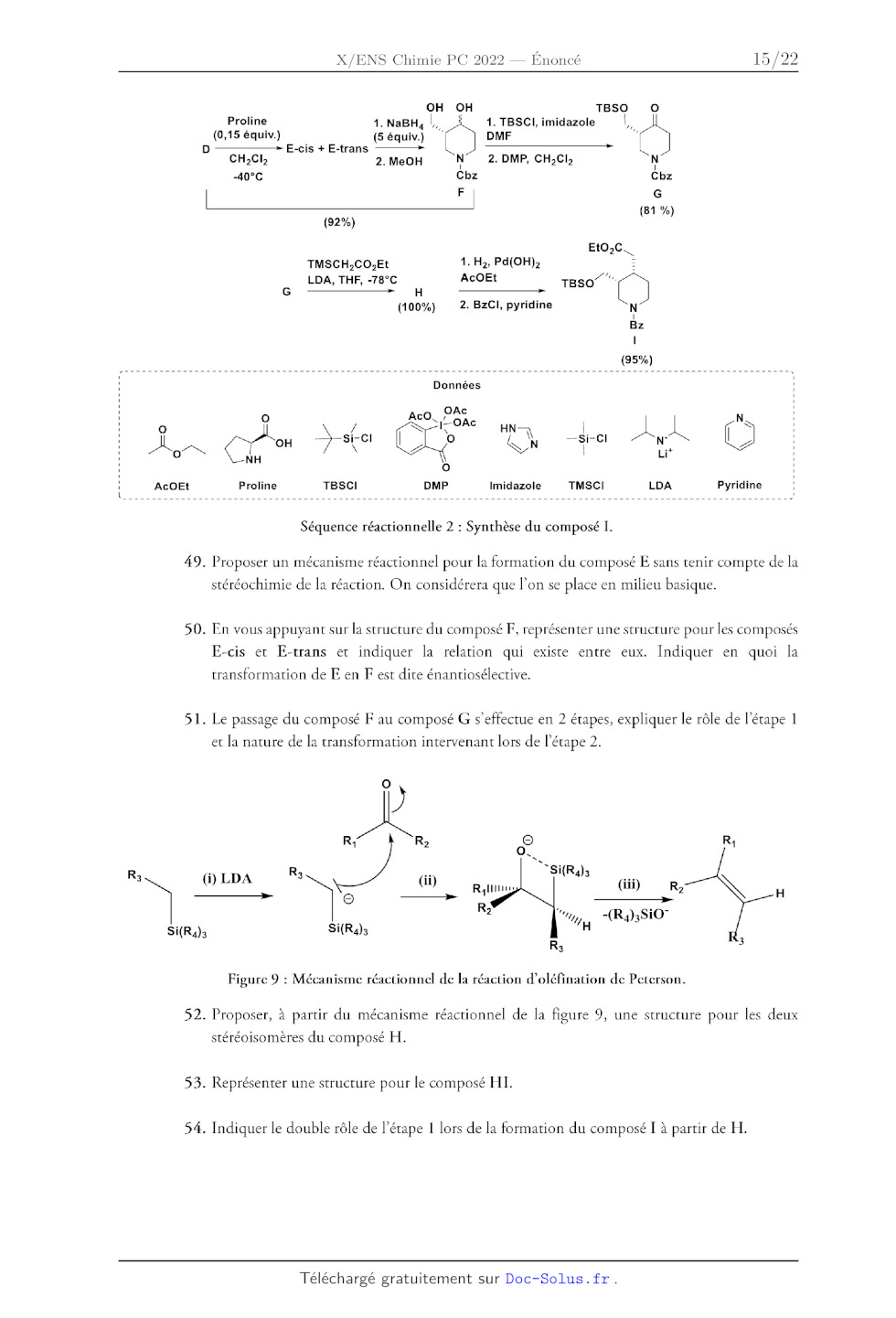
Oo
AL
49.
50.
51.
OH OH TBSO
Proline 1. NaBH, |. 1. TBSCI, imidazole |,
(0,15 équiv.) (5équiv.) DMF :
-- E:-cis + E-trans -- EE
CH,Cl 2. MeOH NT 2. DMP, CH;CI; N
-40°C Cbz Cbz
(81 %)
(92%)
EtO,C\
TMSCH,CO.Et 1. H;, Pd(OH); Es
LDA, THF, -78°C AcOEt reso""(")
GG -------- H
(100%) 2. BzCI, pyridine N
|
Bz
ll
(95%)
Données
. AcO, Pac N
st cl Fe" PE 4 cl À À | |
| --Si- N°
NH h è ÇA Lit 7
Q |
Proline TBSCI DMP Imidazole TMSCI LDA Pyridine !
Séquence réactionnelle 2 : Synthèse du composé I.
Proposer un mécanisme réactionnel pour la formation du composé E sans tenir
compte de la
stéréochimie de la réaction. On considérera que l'on se place en milieu basique.
En vous appuyant sur la structure du composé F, représenter une structure pour
les composés
E-cis et E-trans et indiquer la relation qui existe entre eux. Indiquer en quoi
la
transformation de E en F est dite énantiosélective.
Le passage du composé F au composé G s'effectue en 2 étapes, expliquer le rôle
de l'étape 1
et la nature de la transformation intervenant lors de l'étape 2.
Rs
Rs (i) LDA Rs NC
Y k
Si(R4)3 à
52.
53.
54.
Figure 9 : Mécanisme réactionnel de la réaction d'oléfination de Peterson.
Proposer, à partir du mécanisme réactionnel de la figure 9, une structure pour
les deux
stéréoisomères du composé H.
Représenter une structure pour le composé HI.
Indiquer le double rôle de l'étape 1 lors de la formation du composé I à partir
de H.
15
Le composé L est synthétisé à partir du composé Î selon la séquence
réactionnelle 3 décrite ci-après. La
formation du composé L est réalisée en présence de AD-mix-$ dont la structure
est donnée en annexe 4. Le
spectre RMN du proton du composé K majoritaire (diastéréoisomère E) dans CDCHL
est le suivant : 8,75
(d, / = 4,4 Hz, 1H), 8,09 (d, 7 = 9,3 Hz, 1H), 7,38 (m, 7H), 7,29 (d, / = 2,9
Hz, 1H), 7,19 (d, / = 15,6 Hz,
1H), 6,48 (dt, 7 = 15,6, 7,6 Hz, 1H), 4,32 (m, 1H), 4,18 (m, 1H), 4,00 (s, 4H),
3,91 - 3,62 (m, 2H), 3,51
- 3,11 (m, 2H), 2,48 (m, 1H), 2,23 - 2,08 (m, 2H), 1,95-1,63 (m, 2H), 1,01 -
0,50 (large, 9H), 0,06 (large,
CH). s signifie singulet, d doublet, t triplet et m multiplet.
T IQ
7 1. NaBH,, MeOH, 0°C AD-mix-8
TEso (© 2. MsCI, pyridine 2294 t-BuOH 9) T
LDA, THF, -78°C N O\ 3. t-BuOKTHF : : «
(85%)
Bz J
(98%) (20%)
Séquence réactionnelle 3 : Synthèse du composé L. MsCI correspond au chlorure
de méthanesulfonyle
CH:SO:CI.
55. (*) Le passage du composé J au composé K s'effectue en 3 étapes qui peuvent
être suivies
chacune par spectroscopie IR. Analyser, en s'aidant de l'annexe 2, ces 3 étapes
en reproduisant
et remplissant le tableau ci-dessous.
Étape Nature ou rôle Bande(s) à suivre en IR Évolution de la
(zone en cm!) (des) bande(s) IR
Exemple : Nature (ex : oxydation, Liaison C-C Disparition /
X acido-basique, cyclisation....) (1000 - 1250 cm'!) Apparition /
Rôle (ex : protection, Déplacement
déprotection, activation...)
1
2
3
56. Représenter la structure du stéréoisomère du composé K majoritairement
obtenu.
57. Indiquer le(s) signal (signaux) RMN du proton qui permet(tent) de conclure
sur le
stéréoisomère Æ du composé K majoritairement obtenu. Justifier leur
multiplicité et indiquer
comment ce(s) signal (signaux) se trouve(nt) modifié(s) dans le cas du
stéréoisomère Z.
Finalement, une série de réactions à partir du composé L conduit à l'obtention
de la (-)-quinine selon
la séquence réactionnelle 4 ci-dessous. Le composé O est formé en 2 étapes à
partir du composé M. Le
composé formé à l'issue de la première étape est noté MO.
16
L
------------------------
58.
59.
GO.
61.
62.
63.
64.
1. MeC(OMe);
PPTS, CH,CI 1. DIiBAH, Toluène
-78°C
--------------------------
2. DMF, reflux
7,
2. TMSCI, CH,Cl) TBSO ()
3. K,CO:, MeOH ï 0.
|
Bz M
(90%)
(85%)
1. Ac20, Et;N
DMAP, THF 1. Oxydation
2. Ph;PCH;Br, t-BuOK (-quini
o 2.H M F . Ph:P r, t-BUOK, THF
. HCI (5M) (78%) 3 3 Q
MeOH
3. KCO:, MeOH (56%)
Séquence réactionnelle 4 : Dernières étapes de la synthèse de la (-)-quinine Q.
Représenter la structure du composé MO.
Proposer un mécanisme réactionnel pour rendre compte de la formation du composé
©.
Représenter la structure du composé P.
Indiquer le nom et le rôle des étapes 1 et 2 permettant de former le composé P.
Proposer des réactifs pour la première étape de la formation de la (-)-quinine
à partir du
composé P.
Donner le nom de la réaction mise en jeu dans l'étape 2 lors de la formation de
la (-)-quinine
à partir du composé P.
(*) Indiquer les étapes (ex : À > AB ou B > C) qui induisent la stéréochimie
des carbones
stéréogènes de la (-)-quinine. Spécifier l'unique étape de la synthèse de la
(-)-quinine qu'il est
nécessaire de changer pour obtenir la molécule de (+)-quinidine au lieu de la
(-)-quinine et
proposer un réactif pour obtenir la (+)-quinidine.
17
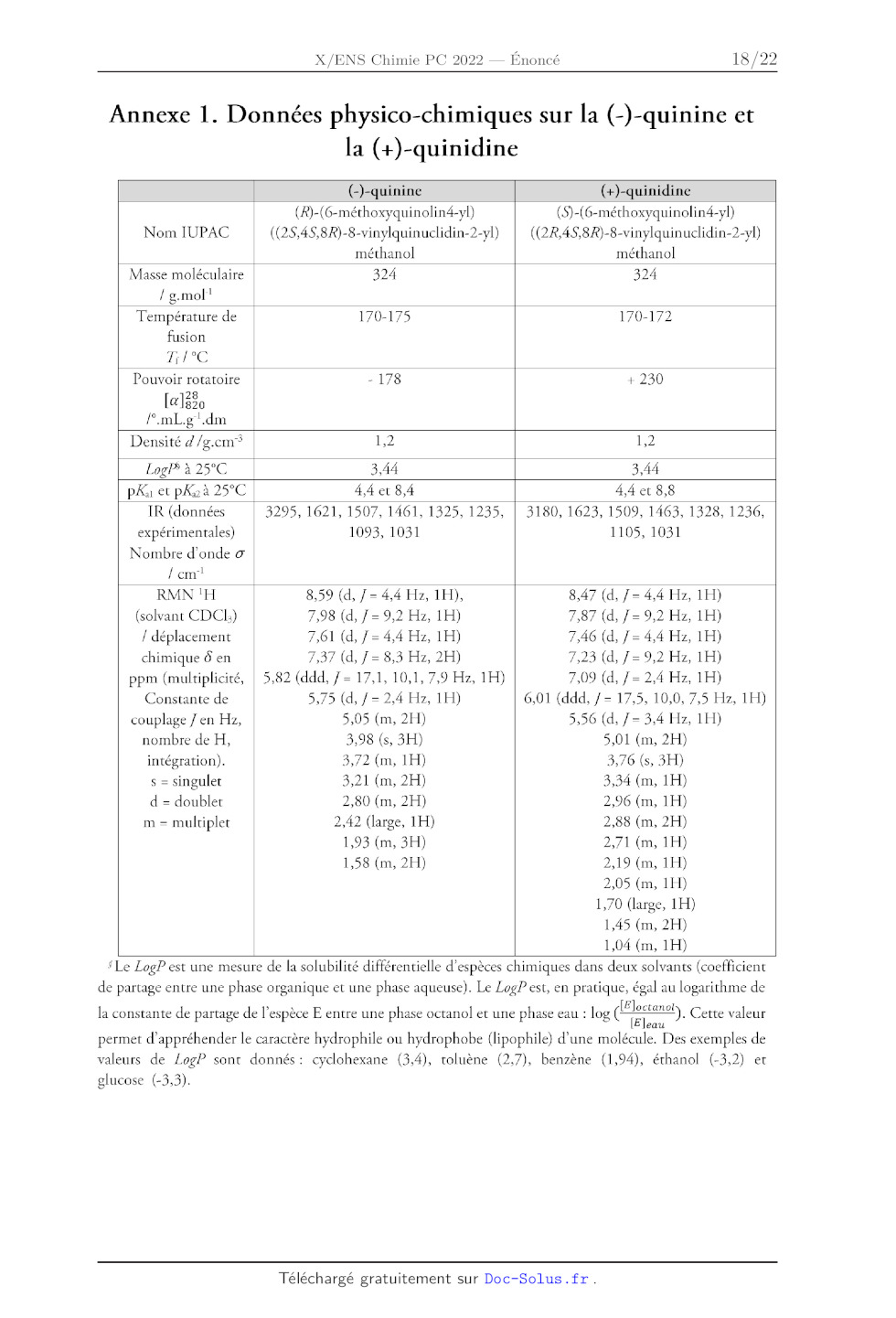
Annexe 1. Données physico-chimiques sur la (-)-quinine et
la (+)-quinidine
(-)-quinine (+)-quinidine
(R)-(6-méthoxyquinolin4-yl) (S)-(6-méthoxyquinolin4-yl)
Nom IUPAC ((2S,45,8R)-8-vinylquinuclidin-2-yl)
((2R4S,8R)-8-vinylquinuclidin-2-yl)
méthanol méthanol
Masse moléculaire 324 324
l g.mol'
Température de 170-175 170-172
fusion
TH °C
Pouvoir rotatoire - 178 + 230
[æ]820
l°.mL.g'.dm
Densité d/g.cm* 1,2 1,2
LogPi à 25°C 3,44 3,44
pXu et pk à 25°C 4,4 et 8,4 4,4 et 8,8
IR (données 3295, 1621, 1507, 1461, 1325, 1235, | 3180, 1623, 1509, 1463, 1328,
1236,
expérimentales) 1093, 1031 1105, 1031
Nombre d'onde &
l'em'!
RMN 'H 8,59 (d, / = 4,4 Hz, 1H), 8,47 (d, / = 4,4 Hz, 1H)
(solvant CDCH) 7,98 (d, 7 = 9,2 Hz, 1H) 7,87 (d, 7 = 9,2 Hz, 1H)
| déplacement 7,61 (d, j = 4,4 Hz, 1H) 7,46 (d, j = 4,4 Hz, 1H)
chimique Ô en 7,37 (d, J = 8,3 Hz, 2H) 7,23 (d, / = 9,2 Hz, 1H)
ppm (multiplicité, | 5,82 (ddd, / = 17,1, 10,1, 7,9 Hz, 1H) 7,09 (d, / = 2,4
Hz, 1H)
Constante de 5,75 (d, 7 = 2,4 Hz, 1H) 6,01 (ddd, j = 17,5, 10,0, 7,5 Hz, 1H)
couplage / en Hz, 5,05 (m, 2H) 5,56 (d, 7 = 3,4 Hz, 1H)
nombre de H, 3,98 (s, 3H) 5,01 (m, 2H)
intégration). 3,72 (m, 1H) 3,76 (s, 3H)
s = singulet 3,21 (m, 2H) 3,34 (m, 1H)
d = doublet 2,80 (m, 2H) 2,96 (m, 1H)
m = multiplet 2,42 (large, 1H) 2,88 (m, 2H)
1,93 (m, 3H) 2,71 (m, 1H)
1,58 (m, 2H) 2,19 (m, 1H)
2,05 (m, 1H)
1,70 (large, 1H)
1,45 (m, 2H)
1,04 (m, 1H)
SLe ZogP est une mesure de la solubilité différentielle d'espèces chimiques
dans deux solvants (coefficient
de partage entre une phase organique et une phase aqueuse). Le LogP est, en
pratique, égal au logarithme de
etant
la constante de partage de l'espèce E entre une phase octanol et une phase eau
: log ( ). Cette valeur
permet d'appréhender le caractère hydrophile ou hydrophobe (lipophile) d'une
molécules Des exemples de
valeurs de ZLogP sont donnés: cyclohexane (3,4), toluène (2,7), benzène (1,94),
éthanol (-3,2) et
glucose (-3,3).
18
Annexe 2. Données spectroscopiques (IR et RMN)
Table IR, en cm'.
Liaison Groupe d'atomes Fonction ou famille Nombre d'onde 6 Intensité
caractéristique (cm)
O-H (libre) C-OH Alcool 3580-3670 Forte
O-H (lié par liaison H) C-OH Alcool 3200-3400 Forte
Carbonyle -COOH Acide carboxylique 3200-3400 Forte
N-H C-NH- Amine, Amide 3100-3500 Moyenne
C-H Cycle benzénique Composés 3030-3080 Moyenne
CH; aromatiques
Alcane 2810-3000 Forte
Alcène 3000-3100 Moyenne
C=0 Carbonyle Aldéhyde, cétone 1650-1730 Forte
Carbonyle Acide 1680-1710 Forte
CO-O-C Ester 1700-1740 Forte
CO-N Amide 1650-1730 Forte
C=C Alcène 1625-1680 Moyenne
C-0 Alcool, acide, ester 1050-1450 Forte
C-C Alcane 1000-1250 Forte
Table de déplacements chimiques en RMN du proton, en ppm.
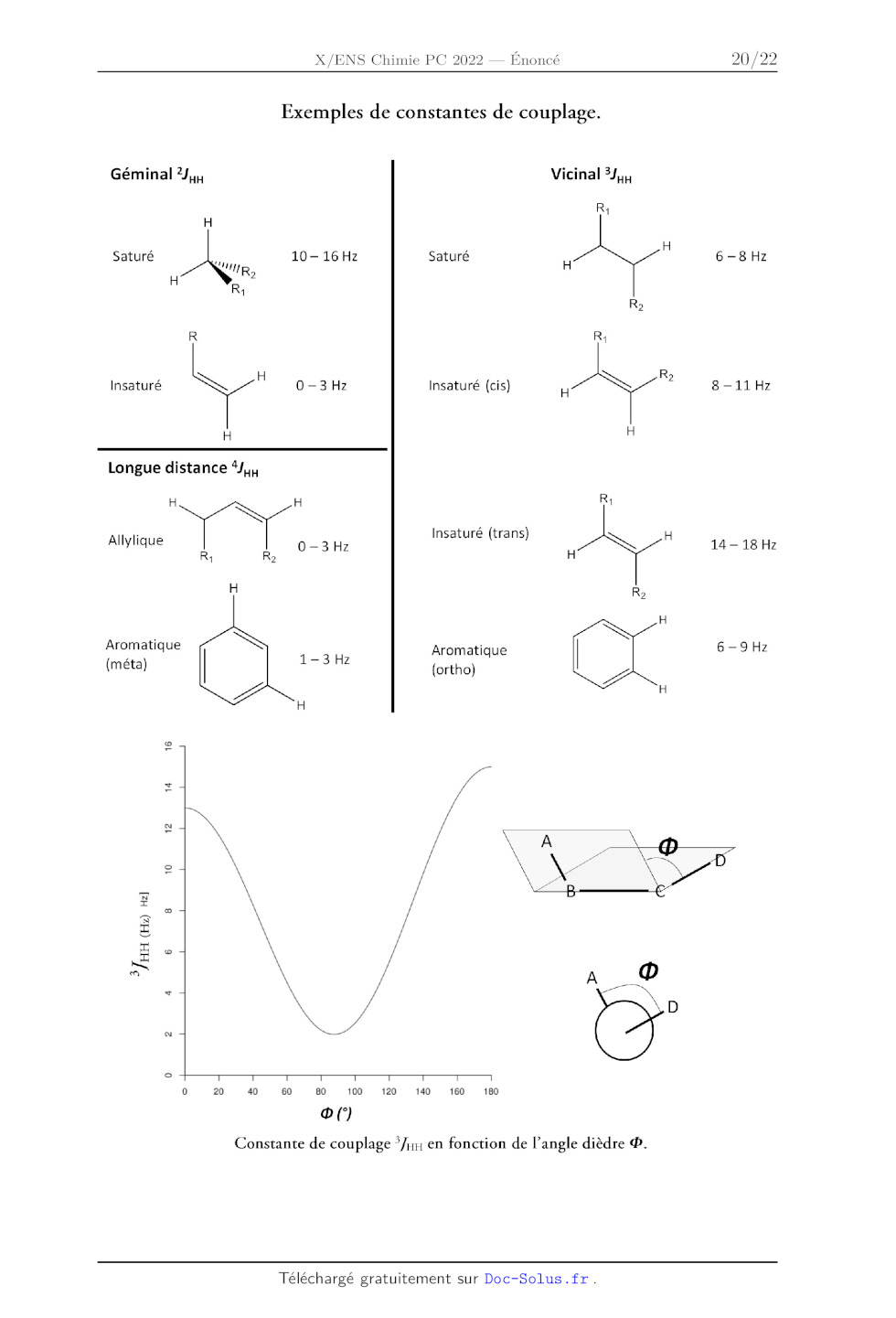
CC) RRRSSSSRRRRRRRERERREREEREREEREEEEEREREEERERRE
8-04 RRSRNININININININIYIYIYFSSRSEREREEKEEEREREEEEEEEERREREERERES]
us-- »
EN US |
R--sH
SJ R- NH" R-NE KKK
KKKKKKKKKKKKKKKK1 e
-- KKKK--CH=CH-- (cycle)
EEEEEEEEQFFFFFFFFFFFFFFF'FFF --CH=CH--
KKKKKKKKKKKKKK > C=CH
SKK --C=CH
R-CH-NT
SSH KW CH
O0
CH-X
Lo KKKKKKKKKKKKKKKKKKK
NS ET
ZC-CH---N0, SS ÉÉNNNSSZC-CH-NT
>c- 0 RRKKININSS SSW>=C-CH-C--
--CH--0--(ye)SS --CH-- RSS ASS
Sec CC R :
DC-CH- CC #
as
FIN SNS TS
III HNLRSISISSS
RÉINONS,"\NINKS -- on Ho RSS
SE 0
oO
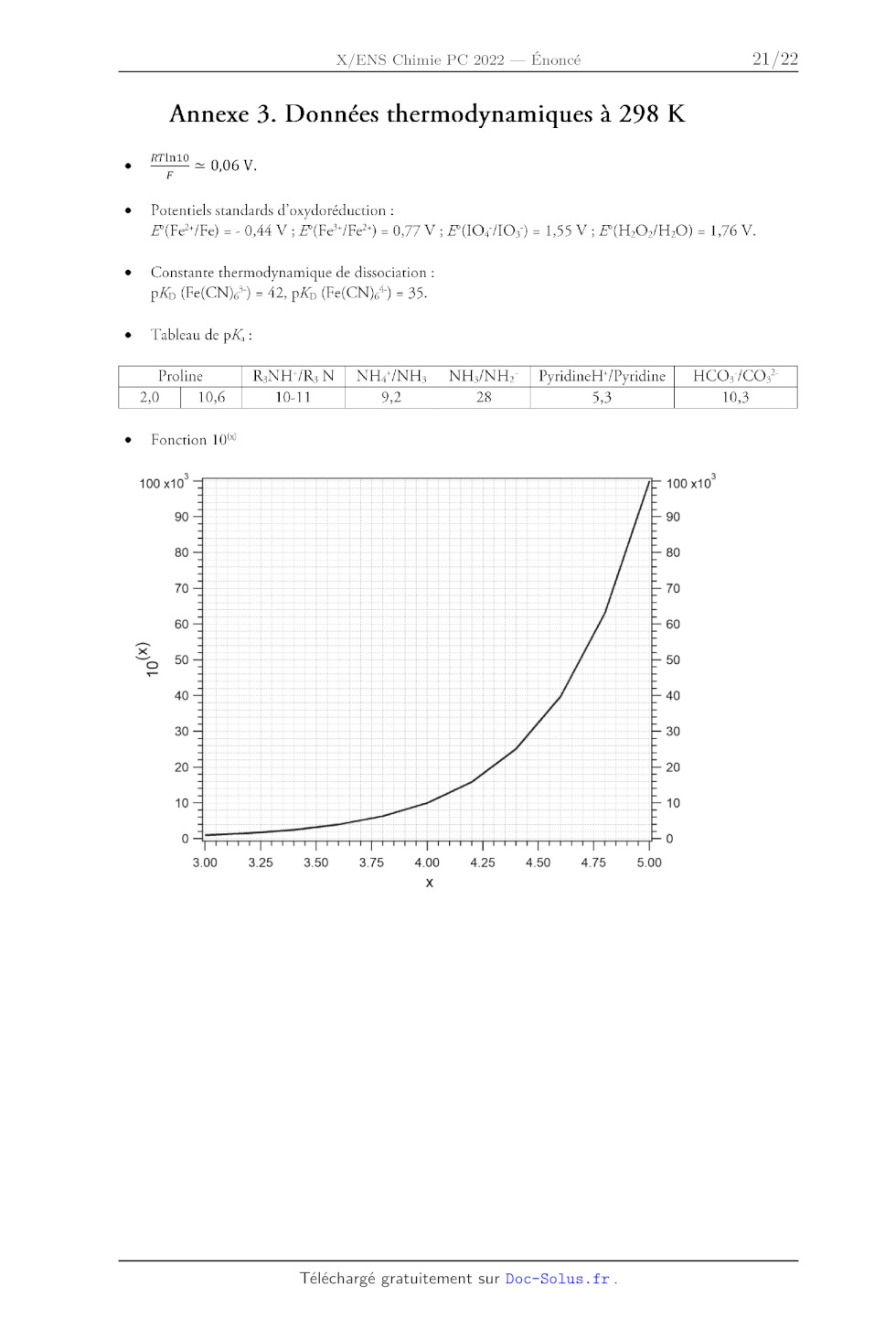
CH;-C=C< CHyS-- | UE SSS CH-É-0-- o CH-G-X Scu;-c= 10.0 9,0 CH-Si= ÔPPM 8,0 7,0 6,0 5,0 4,0 3,0 2,0 1,0 0 19 Exemples de constantes de couplage. Géminal 2J,,, Vicinal 5J,4, R: , , H Saturé 10-16Hz Saturé 6-8 Hz Y.e H Re R: R Insaturé y 0 - 3 Hz Insaturé (cis) A ? 8 --11 Hz H Longue distance Longue distane e R: NK Allylique 0-34 Insaturé (trans) | NS H 1418 R: H Aromatique : 6---9Hz né 1-3 Hz Aromatique (ortho) H 16 14 12 10 3], HH (Hz) Ha] 6 0 20 40 60 80 100 120 140 160 180 oe( Constante de couplage */nn en fonction de l'angle dièdre ®. 20 Annexe 3. Données thermodynamiques à 298 K RTIn10 _ 0,06 V. Potentiels standards d'oxydoréduction : F(Fe*/Fe) = - 0,44 V ; F(Fe*/Fe?) = 0,77 V ; E(1O4/10;) = 1,55 V ; £(H:02/H20) = 1,76 V. Constante thermodynamique de dissociation : pXs (Fe(CN)é) = 42, pXp (Fe(CN)*) = 35. Tableau de pX, : Proline RNHY/R N°) NHS/NH3 | NH3/NH> | PyridineH'/Pyridine HCO;/CO;-
2,0 10,6 10-11 9,2 28 5,3 10,3
Fonction 10%
100 x10° 100 x10°
90 90
80 80
70 70
60 60
= 50 50
40 40
30 30
20 20
10 10
0 0
300 3.25 3.50 3.75 4.00 4.25 450 4.75 5.00
x
21
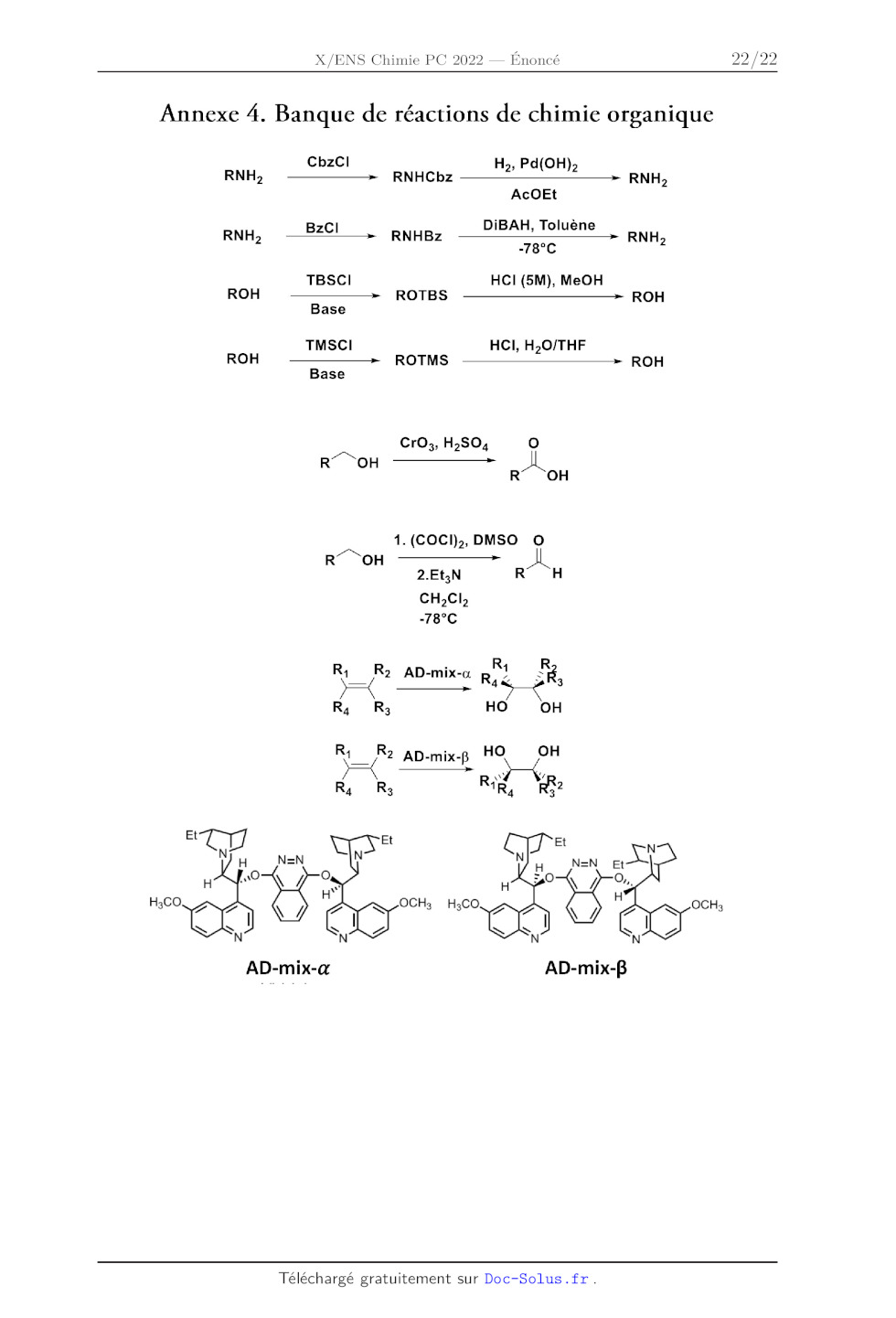
Annexe 4. Banque de réactions de chimie organique
CbzCl H,, Pd(OH)
RNH; RNHCbz 7" 2, RH,
AcOEt
DiBAH, Toluène
RNHy --B2CT , RaBz "TT RNH,
78°C
TBSCI HCI (5M), MeOH
ROH ROTBS ROH
Base
TMSCI HCI, H,O/THE
ROH --"", ROTMS ROH
Base
ROH
R° OH
1. (COCI);, DMSO O
RO OH
2.Et;N RT CH
CH,Cb
-78°C
R R
R R -mix- 1 < 1/92 AD-mix-o RE à Rs R, Rs HO OH Ra R2 AD-mix-5 HO OH Re Ra R3 R4 29